

 PDF
PDF Citation
Citation Print
Print


Introduction
Glioblastoma multiforme (GBM) is the most malignant and common brain tumor and it comprises ~23% of all primary brain tumors in adults. These malignancies are refractory to all the current therapeutic approaches, including surgery, radiotherapy and chemotherapy. Fas (CD95 or APO-1) is a member of the TNF/NGF receptor family, and Fas induces caspase-dependent apoptotic death in various transformed cells (1,2). Fas ligation with natural ligand or agonistic anti-Fas antibody is followed by recruitment of proapoptotic adaptor molecules such as Fas-associated death domain (FADD) to transduce the apoptotic signals through the caspase cascades (3). In some cells, Fas efficiently activates caspase-8 and it subsequently activates caspase-3 or 7, while other types of Fas-induced apoptosis are mediated by cytochrome-C release from the mitochondria and this is inhibited by the over-expression of anti-apoptotic bcl-2 family members (4).
Panax Ginseng is known for its biological and pharmacological activities such as its anti-cancer, anti-aging, anti-inflammatory and anti-oxidant properties in the nervous, immune and circulatory systems (5). These diverse physiological activities of ginseng are mainly mediated by saponin, which is a ginsenoside. Especially, the metabolites of ginsenosides that are formed by enteric bacteria have been focused on for their pharmacological activities. Among them, compound K (C-K) is known to be formed by enteric bacterial fermentation of Rb1, Rb2 and RC, and C-K has been reported to suppress tumor metastasis and inflammatory responses (6,7). Another ginsenoside Rh2, a metabolite of Rg3, is also known for its tumor suppression by inducing apoptosis or retarding growth signals (8).
We have previously shown that human malignant astrocytoma cells are quite resistant to Fas-induced apoptosis even though these cells express functional Fas on their surface (2,9). Even though the role of reactive oxygen species (ROS) has been controversial in terms of receptor-induced apoptosis, it has been shown that the inhibition of receptor-induced ROS generation augmented the Fas-mediated apoptosis in human astrocytoma cells, and this suggests the anti-apoptotic role of ROS. In this study, we investigated the molecular mechanisms that are responsible for killing of tumor cells by pro-apoptotic ginsenosides and the augmentation of Fas-induced cell death in human astrocytoma cells.
Go to : 
Materials and Methods
1. Cell culture
Human astrocytoma CRT-MG cells were grown in RPMI 1640 medium that was supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin G (100 U/ml), streptomycin (100 µg/ml) and L-glutamine (2 mmol/L) in a 5% CO2 incubator at 37℃, as previously described (10). Other human astrocytoma cell lines, U251-MG and U87-MG cells, were maintained in Dulbecco's modified Eagle media (JBI, Korea) that was supplemented with 10% FBS and penicillin G (100 U/ml). Primary human fetal astrocytes were obtained from therapeutically aborted fetal brains and they were maintained in Dulbecco's modified Eagle media that was supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin G (100 U/ml) and 1% nonessential amino acids (Gibco-BRL, Grand Island, NY), as previously described (11).
2. Reagents
Ginseng saponin ginsenosides (F1, Ro, Rc, Re, Rd, Rf, C-K, Rh2, Rg1, Rg2, Rg3, Rb1 and Rb2) were obtained from KT&G (Daejeon, Korea). N-acetyl cysteine (NAC), 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) and diphenyl iodonium (DPI) were all purchased from Sigma (St. Louis, MO). Dichlorodihydrofluorescein diacetate (DCF-DA) and tetramethylrho-damine ethyl ester (TMRE) were purchased from Molecular Probe (Eugene, OR). An agonistic IgM type anti-Fas antibody (CH-11) was obtained from Upstate Biotechnology (Lake Placid, NY). Human recombinant TNF-α and Fas ligand were purchased from R&D Systems (Minneapolis, MN). Caffeic acid phenethyl ester (CAPE) and SB202190, SP100625 and U0126, which are pharmacological inhibitors of p38 MAPK, JNK and ERK, respectively, were obtained from Calbiochem (La Jolla, CA).
3. Measurement of the intracellular ROS levels
To detect intracellular ROS, an oxidation-sensitive probe 2, 7-dichlorofluorescein-diacetate (DCF-DA) was used as previously described (9). To study the time course of Fas-mediated ROS production, the cells were incubated with 2 µmol/L of DCF-DA for 10 min and then they were treated with CH-11 (500 µg/L) for varying time periods (0~60 min). To investigate the effect of NAC and CAPE on Fas-induced ROS production, the CRT-MG cells were maintained in serum-free media for 16 h, incubated in the absence or presence of these inhibitors for 1 h and then they were treated with CH-11 (500 µg/L) for an additional 15 min. The cellular fluorescence was measured using an inverted epifluorescence microscope (Zeiss, Germany).
4. Measurement of cell death
Cell death was determined by staining with Annexin V (PharMingen), which is a 35.8-kDa protein that has a strong affinity for phosphatidylserine (12). The cells were washed with PBS, trypsinized, suspended in 200 µL of binding buffer and stained with 0.5 ng of Annexin V-fluorescein isothiocyanate (FITC) and 2.5 ng of propidium iodide (PI). Ten thousand cells were analyzed with using a FACStar (Becton Dickinson, Mountain View, CA) within 30 min after staining. Cell death was defined as those cells that were positive for Annexin V and/or PI. The mitochondrial transmembrane potential (Δψm) was assessed after staining with TMRE. The cell suspension was incubated in 2 µmol/L of TMRE for 30 min at room temperature and then it was analyzed with a FACStar.
The MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay was also used to determine cell viability, as described previously (13). Briefly, MTT is a yellow, water-soluble, tetrazolium salt. Metabolically active cells are able to convert this dye into a water-insoluble, dark-blue formazan by reductive cleavage of the tetrazolium ring. The formazan crystals can then be dissolved and quantified by measuring the absorbance of the solution at 595 nm, and the resultant value is related to the number of living cells.
5. Subcellular fractioning
The cells were washed with ice-cold PBS and then vortexed in the hypotonic buffer solution that contained 500 mmol/L sucrose, protease inhibitors and Na3VO4 (10 mmol/L Hepes, 2 mmol/L MgCl2, 25 mmol/L KCl, 0.5 mmol/L EDTA, 0.5 mmol/L EGTA). Then NP-40 was added to the final concentration of 0.05%, and the mixture was centrifuged at 15,000g for 15 min. The supernatants were assayed for the cytosolic fractions, and the precipitants were analyzed for the mitochondrial fractions.
6. Immunoblot analysis
Immunoblot analysis for caspases was performed as previously described (14). The cell lysates (20 µg) were electrophoresed in 10% SDS-PAGE gels, the proteins were transferred to nitrocellulose membranes and these were probed with antibodies against human caspase-3, PARP, p38 MAPK, phosphor-specific p38 MAPK and β-actin (Cell Signaling, Danvers, MA). The blots were developed by chemiluminescence (AbFrontier, Korea).
7. Statistical analysis
The data is presented as means ±SDs. Comparisons between two different samples were analyzed by the Student t-test, and comparisons between more than three different samples were done by ANOVA (analysis of variance) with applying Tukey's post-hoc test to the significant main effects or interactions (SPSS 12.0K for Windows, SPSS, Chicago, IL).
Go to :
Results
1. Cell death of the malignant astrocytoma cells by proapoptotic ginsenosides
We screened thirteen ginsenosides for their anti-cancer effect on human malignant astrocytoma cells (CRT-MG, U87-MG and U251-MG cells). Among the 13 ginsenosides, compound K (C-K) and Rh2 induced marked time-dependent cytotoxicity even at a concentration as low as 25 mg/L, while the other ginsenosides showed marginal cytotoxic effects at a concentration as high as 50 mg/L (Fig. 1A). The cytotoxic effect of C-K and Rh2 was only observed for the transformed tumor cells, but not for the primary cultured human astrocytes even at higher concentrations of C-K and Rh2 (Fig. 1B). Interestingly, the cells treated with C-K showed vacuolar changes even with using concentrations that had little toxicity. To test what type of cell death was induced by ginsenosides, the cells treated with the ginsenosides were stained for Annexin V and propidium iodide (Fig. 1C). As expected, treatment with C-K and Rh2 induced typical apoptotic cell death, which was confirmed by the positive staining with Annexin V. Pretreatment with a broad-spectrum caspase inhibitor z-VAD-Fmk markedly reduced the ginsenoside induced cell death, confirming that ginsenosides induced caspase-dependent apoptotic cell death in the malignant astrocytoma cells. Since it has been reported that the mitochondrial intermembrane potential (Δψm) is selectively decreased during the activation of the apoptotic cascades (15), we further investigated whether treatment with ginsenosides affected the Δψm by staining with TMRE (Fig. 1D). In concordance with the Annexin V staining results, treatment with C-K and Rh2 induced a rapid depolarization of the Δψm, and this was nearly completely reversed by pretreatment with z-VAD-fmk. These results clearly indicate that the ginsenosides C-K and Rh2 induced caspase-dependent apoptotic cell death in human malignant astrocytoma cells.
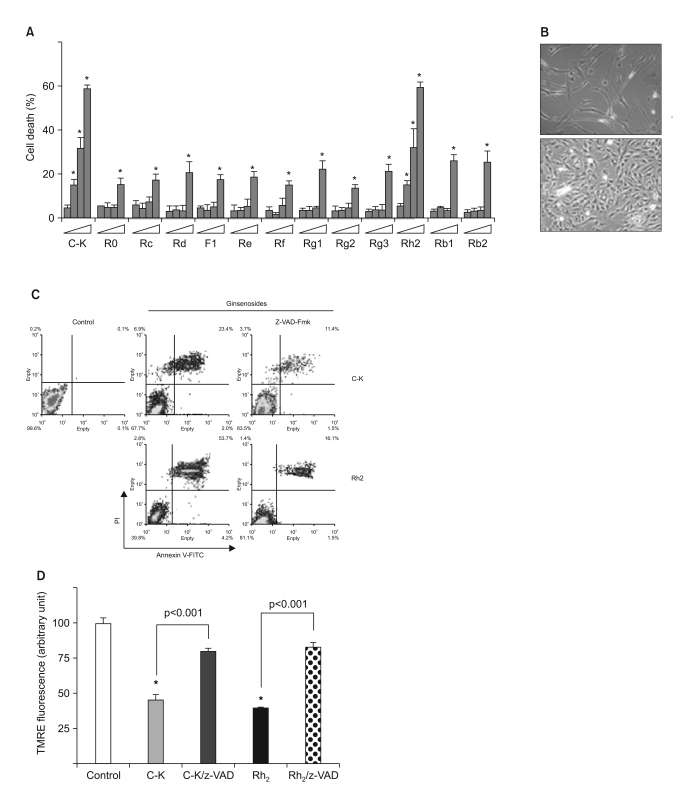 | Fig. 1Caspase-dependent apoptosis by the ginsenosides C-K and Rh2 in human astrocytoma cells. (A) The cells were treated with varying doses (0~50 mg/L) of 13 different ginsenosides, and the cell death was determined by MTT assay. *, significantly different from the control sample without any treatment (p<0.01). (B) Human primary cultured astrocytes and CRT-MG cells were treated with 25 mg/L C-K for 6 h. (C) The cells were incubated in the absence or presence of z-VAD-Fmk (10 mmol/L) for 1 h, and then they were treated with C-K or Rh2 for an additional 24 h, and the cell death was next determined after staining with Annexin-V-FITC and PI by FACS analysis. (D) The cells were incubated in the absence or presence of z-VAD-Fmk (10 mmol/L) for 1 h; they were next treated with C-K or Rh2 for an additional 24 h and then they were stained with TMRE. *, significantly different from the control sample (p<0.001).
|
2. The signaling pathways involved in ginsenosides-induced apoptosis
We next investigated the signaling pathways that are responsible for the ginsenoside-induced apoptotic cell death. Since we have observed that inhibition of caspases significantly suppressed the ginsenoside-induced cell death, we first tested the involvement of specific caspases by performing western blot analysis (Fig. 2A, B). Caspase-3 is known as the common executioner of apoptotic cell death, and it was cleaved into active fragments (17/19 kDa) in a time-dependent manner by treatment with C-K or Rh2. We observed the proteolytic cleavage of caspase-3 as early as 12 h after treatment with ginsenosides, while the maximal cleavage was observed 24 h after treatment. The proteolytic cleavage of PARP (poly-ADP ribose polymerase), a well known cellular target of activated caspase-3, was also observed in a time-dependent manner after treatment with C-K (Fig. 2B). To further investigate the molecular mechanisms of ginsenoside-induced apoptosis, we further examined the cytosolic translocation of cytochrome-C (Fig. 2C). Treatment with C-K induced a marked translocation of mitochondrial cytochrome-C into the cytosolic fractions. These results collectively suggest that ginsenosides induce apoptotic cell death via the intrinsic mitochondrial pathways.
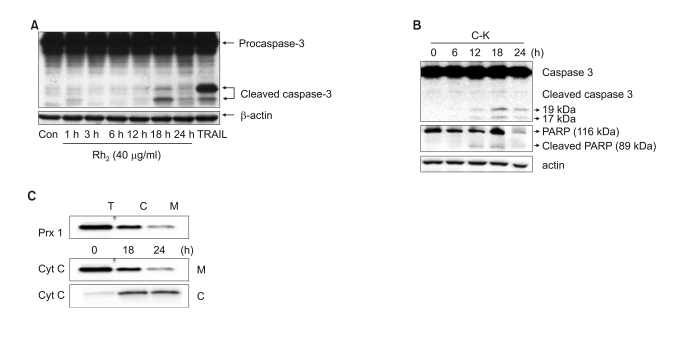 | Fig. 2The involvement of caspase-3 and cytochrome C release in ginsenosides-induced cytotoxicity. (A) CRT-MG cells were incubated with Rh2 for varying time periods, and the cell lysates were subjected to western blot analysis for caspase-3 and b-actin. (B) CRT-MG cells were incubated with C-K for varying time periods, and the cell lysates were subjected to western blot analysis for caspase-3, PARP and b-actin. (C) The total cell lysates (T) were divided into the cytosolic fraction (C) and the mitochondrial fraction (M), and peroxiredoxin-1 was examined to test the separation efficiency (upper panel). The mitochondrial fraction (M) and the cytosolic fraction (C) were tested for their cytochrome-c levels.
|
We further tried to investigate the intracellular signaling pathways that are responsible for the activation of the intrinsic apoptotic pathways. We examined the involvement of the mitogen activated kinase (MAPK) pathways for controlling the ginsenoside-induced apoptosis by using various specific pharmacological inhibitors for ERK1/2, p38 and JNK. Pretreatment with the p38-specific inhibitor significantly suppressed C-K-induced cell death, while the pretreatment with ERK or JNK inhibitors had no effect on C-K-induced cell death (Fig. 3A). The protective effect of p38 inhibitor was not observed for the Rh2-induced cell death. The protective effect of p38 inhibitor on the C-K-induced cell death was also confirmed by Annexin V staining (data not shown). We next tested the involvement of p38 MAPK in the C-K-induced cell death by performing western blot analysis. As expected, treatment with C-K induced a time-dependent phosphorylation of p38 MAPK, which was sustained up to 18 h after treatment (Fig. 3B).
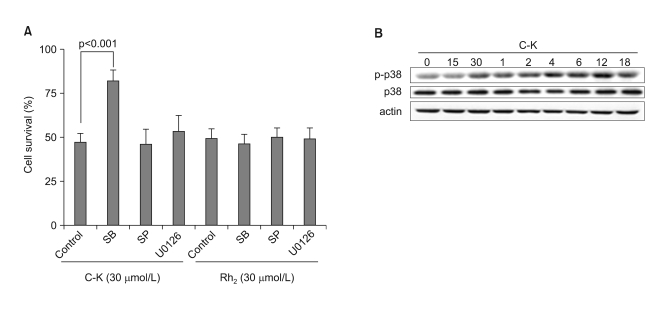 | Fig. 3The involvement of p38MAPK in C-K-mediated cell death. (A) CRT-MG cells were incubated in the absence or presence of various MAPK inhibitors for 1 h, and then they were treated with C-K or Rh2 for an additional 24 h, and cell death was measured by MTT assay. (B) The cells were treated with C-K (25 mg/L) for varying time periods, and the total cell lysates were subjected to western blot analysis for p38 MAPK, phospho-p38 MAPK and β-actin.
|
3. The synergistic effect of ginsenosides on Fas-induced cell death
We next investigated whether pro-apoptotic ginsenosides can sensitize Fas-resistant astrocytoma cells to Fas-induced cell death by performing MTT assay. To test the effect of C-K on Fas-mediated cell death, the CRT-MG cells were incubated in the absence or the presence of varying concentrations of C-K for 1 h and then the cells were treated with increasing doses of CH-11 anti-Fas antibody for 24 h. As CRT-MG cells are known to be resistant to Fas-induced cell death (16), the cells did not undergo cell death upon Fas ligation in the absence of C-K (Fig. 4A). Yet pretreatment with increasing doses of C-K dramatically increased the sensitivity to Fas-mediated cytotoxicity in a dose-dependent manner. A similar synergistic effect on Fas-mediated cell death was observed by pretreatment with Rh2 (data not shown). The synergistic effect of ginsenosides with fas ligation was also confirmed by Annexin V staining and the mitochondrial depolarization (Fig. 4B, C). We also observed that the inhibition of caspases suppressed the synergistic cytotoxicity induced by ginsenosides (data not shown).
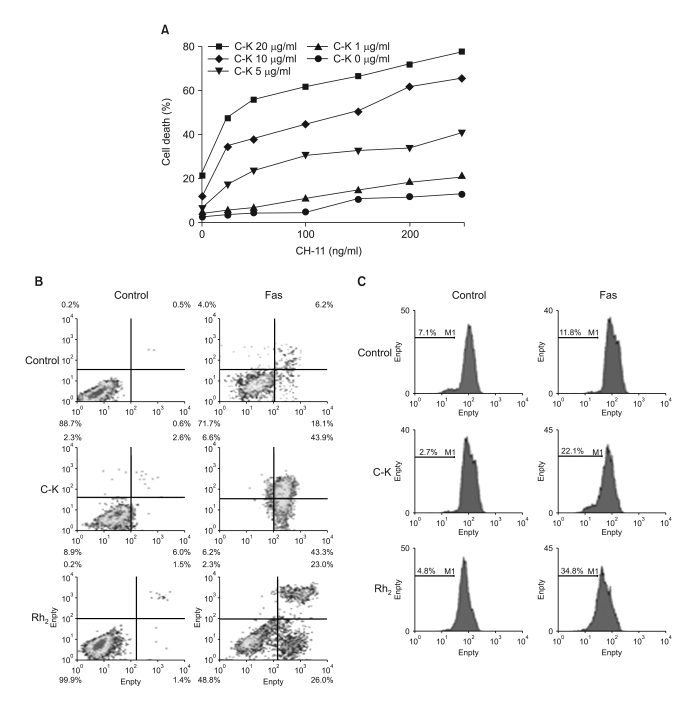 | Fig. 4Ginsenosides augment fas-mediated cell death. (A) The cells were incubated in the absence or presence of varying doses of C-K for 1 h, and then they were treated with CH-11 for an additional 24 h, and the cell death was measured by MTT assay. (B) The cells were incubated in the absence of presence of C-K or Rh2 for 1 h, and then they were treated with CH-11 for an additional 24 h, and the cell death was measured after staining with Annexin-V and PI by FACS analysis. (C) The cells were incubated in the absence or presence of C-K or Rh2 for 1 h, and then they were treated with CH-11 for an additional 24 h, and the Δψm was measured after staining with TMRE.
|
We have previously reported that ROS scavengers such as NAC and CAPE enhanced Fas-induced cell death in a caspase-dependent manner (9). Since various ginsenosides are known for their effective ROS scavenging activities, we tested the possibility that C-K and Rh2 can enhance Fas-induced apoptosis in a ROS-dependent manner. As reported previously (9), we were able to confirm that Fas ligation induced a time-dependent increase of the intracellular ROS levels (Fig. 5A). Because the maximal induction of the intracellular ROS levels was observed after 15 min of Fas ligation, we used that time point for the following experiments. In agreement with our hypothesis, the pretreatment with C-K or Rh2 significantly reduced the intracellular ROS levels induced by Fas ligation (Fig. 5B). To confirm whether inhibition of ROS generation can enhance Fas-induced cell death via caspase-3 activation, we further tested the in vitro caspase-3 enzymatic activity of the lysates of the cells treated with CH-11 in the absence or presence of ginsenosides (Fig. 5C). Treatment with the ginsenosides alone had little effect on the caspase-3 enzymatic activity, while the same treatment significantly enhanced the Fas-induced enzymatic activity of caspse-3.
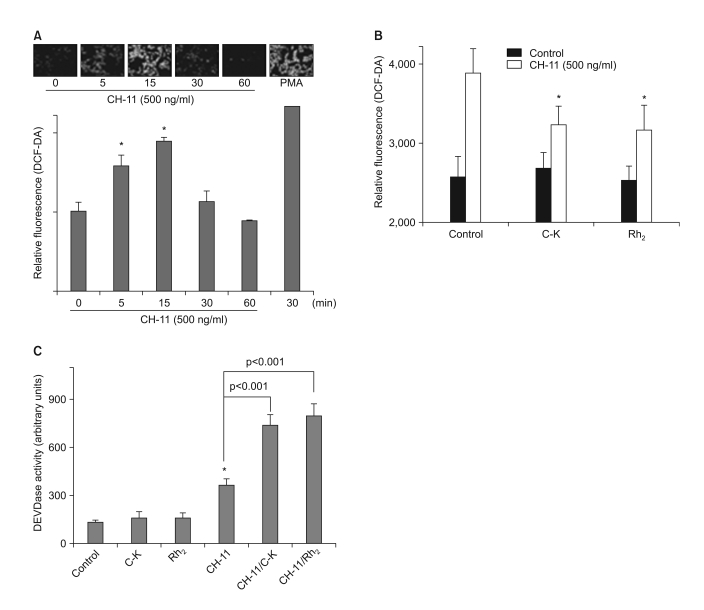 | Fig. 5Ginsenosides Suppress Fas-Induced ROS Generation. (A) The CRT-MG cells were incubated with anti-Fas antibody (CH-11) for varying time periods, and then they were examined for their intracellular ROS levels by staining with DCF-DA. *, significantly different from the control sample (p<0.05). (B) The cells were incubated in the absence or presence of C-K or Rh2 for 1 h, and then they were treated with CH-11 for an additional 30 min, and the intracellular ROS levels were measured. *, significantly different from the control sample treated with CH-11 alone (p<0.01). (C) The cells were incubated in the absence or presence of C-K or Rh2 for 1 h, and then they were treated with CH-11 for an additional 2 h, and the in vitro DEVDase enzymatic activity was measured. *, significantly different from the control sample without any treatment (p<0.01).
|
Go to :
Discussion
Several ginsenosides, and especially C-K and Rg3 as enteric metabolites of Rb1, Rb2 and Rc, have been reported on for their anti-cancer activities that occur through various mechanisms (17). In agreement with these previous reports, we also observed the anti-proliferative effects of C-K and Rh2 on human malignant astrocytoma cells in a caspase- and mitochondria-dependent manner. We have observed that caspase-8, an upstream caspase involved in the extrinsic cell death pathway, is not cleaved in the process of ginsenoside-induced apoptosis (data not shown). Along with the data showing mitochondrial depolarization and cytosolic translocation of cytochrome-C by ginsenosides, these results confirmed the involvement of mitochondria in the ginsenoside-induced cell death of astrocytoma cells.
MAP kinases are the family of protein kinases that includes extracellular signal regulated kinase (ERK), c-Jun N-terminal protein kinase (JNK) and p38 MAP kinase. These protein kinase cascades have been implicated in various cellular responses such as cellular proliferation and death (18). Among them, p38 MAPK is reported to be involved in the tumor cell death induced by various chemotherapeutic agents, and this cell death mainly occurs through p53 activation and mitochondrial depolarization (19). Activation of p38 MAPK is reported to be mediated by the specific upstream kinases MKK3/6 (18). Further research is needed to determine the exact molecular mechanisms that are responsible for p38 MAPK activation by ginsenosides.
Even though we have shown the pro-apoptotic activities of ginsenosides on human malignant astrocytoma cells, the clinical application of these reagents as a single therapy doesn't seem to be promising. We have witnessed that many reagents with wonderful in vitro activities failed to have satisfactory clinical efficacy due to various reasons such as development of resistance mechanisms by tumor cells (20). Combining multiple anti-cancer drugs, and especially those drugs with different modes of action, will dramatically decrease the probability of developing resistance and so enhance the drugs' clinical efficacy for treating malignant tumors. Therefore, the combined treatment of ginsenosides and anti-Fas ligation will not only increase the anti-tumor cytotoxic efficacy, but this will also effectively prevent the occurrence of tumor resistance (Fig. 6).
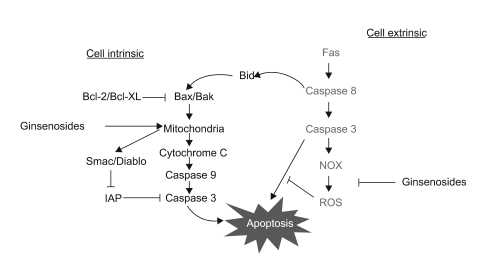 | Fig. 6The proposed model for the anti-tumor activity of ginsenosides and the therapeutic strategies for treating malignant tumors. Ginsenosides induce apoptotic cell death in a mitochondria-dependent manner, while the sensitizing effect of ginsenosides for Fas treatment occurs by ROS inhibition. Since ginsenosides can induce intrinsic cell death and they augment Fas-induced extrinsic cell death, a combined treatment of Fas and ginsenosides might be employed as a useful therapeutic strategy for treating multi-drug resistant malignant astrocytomas.
|
We have previously shown that the inhibition of the Fas-induced ROS response rendered astrocytoma cells susceptible to Fas-mediated apoptosis in a caspase-dependent manner (9). The synergistic effect of ginsenosides on Fas-induced cell death also seems to be related to the anti-oxidant activities of these proapoptotic ginsenosides. These results collectively suggest that the Fas-induced ROS responses might be a negative feedback regulator for the caspase-dependent apoptotic cascades. It has been proposed that endogenous ROS can be anti-apoptotic under certain circumstances (21). Superoxide anion and hydrogen peroxide are known to be generated during oxidative phosphorylation or in response to growth factors and cytokines (22). The role of ROS has been reexamined as being part of the essential intracellular signaling cascades rather than being just a harmful waste product of aerobic respiration. However, its biological function during programmed cell death has been debated. It has been proposed that receptor-mediated generation of ROS is pro-apoptotic because the Fas-induced cell death in various cell types was protected by administering antioxidants (23,24). On the other hand, pro-oxidative conditions have also been shown to be protective against receptor-mediated apoptosis under certain circumstances.
Go to :
Conclusions
Our results demonstrated that the ginsenosides C-K and Rh2 induce tumor-specific apoptotic cell death of human malignant astrocytoma cells via the intrinsic pathways along with the involvement of the mitochondria. Furthermore, we have shown that p38 MAPK is the major intracellular signaling pathway that's responsible for caspase-dependent cell death. We also showed that these pro-apoptotic ginsenosides rendered astrocytoma cells susceptible to Fas-mediated apoptosis in a caspase- and ROS-dependent manner. These results will provide the background for developing more effective anti-cancer strategies against highly malignant brain tumors.
Go to :
 XML Download
XML Download