

 PDF
PDF Citation
Citation Print
Print


Introduction
Desmoplastic small round cell tumor (DSRCT) is a very rare malignancy, which was 1st described by Gerald and Rosai in 1989 (1). DSRCT primarily occurs in adolescent males and develops in the abdominal cavity (2). Although its origin has not been identified, DSRCT is believed to arise from the mesothelium, because it is frequently found in the mesothelial-lined surface.
Typical pathologic findings include abundant desmoplastic stroma and poorly differentiated small cells, which have various differentiations such as epithelial, mesenchymal, or neuronal differentiation (3-5). DSRCT, as well as Ewing sarcoma/PNET (primitive neuroectodermal tumor), has EWS gene translocations (6,7).
Most patients are diagnosed in the advanced stages and have a poor prognosis. Some studies have indicated the possibility of long term survival by means of combination chemotherapy, radiation therapy, and debulking surgery (8-13).
We report the case of a young female patient who was diagnosed with DSRCT and was treated with combination chemotherapy.
Case Report
A 26-year old woman was admitted to our hospital with dyspepsia and abdominal pain lasting for 2 months. She visited the local clinic for dyspepsia and abdominal pain 2 months prior to admission and was examined using esophagogastroduodenoscopy, which at that time revealed no abnormal findings. However, 18 days prior to admission, the patient found a palpable mass located in the right upper quadrant of her abdomen and visited a clinic. On endoscopic examination, there was an encircling mass in the second portion of the duodenum, which was suspected to be an undifferentiated carcinoma. Then, she was referred to our hospital.
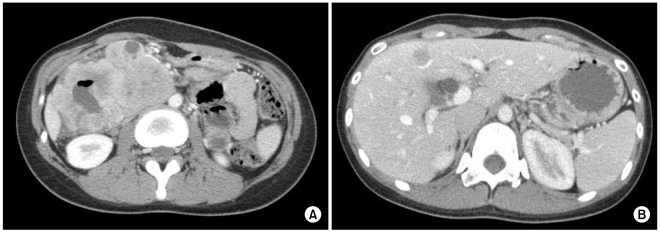
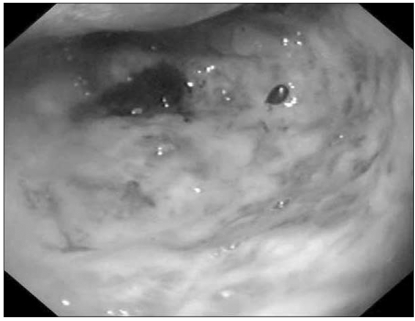
She had an approximately 10 cm-sized palpable and tender abdominal mass in the right upper quadrant on first visit. In addition to the abdominal mass, the patient was diagnosed with iron deficiency anemia. The results of liver function tests were consistent with obstructive jaundice: total bilirubin 2.2 mg/dL (normal range, 0.2~1.2), alkaline phosphatase 452 mg/dL (normal range, 30~115), aspartate aminotransferase (AST) 131 IU/L (normal range, 0~40), alanine aminotransferase (ALT) 294 IU/L (normal range, 0~40), and gammaglutamyl transpeptidase (GGT) 579 IU/L (normal range 11~63). Serum neuron specific enolase level was 26.3 ng/mL (normal range, 0~16.3). Abdominal plain radiography was consistent with mechanical ileus. Abdominal computed tomography (CT) scan revealed wellenhanced abdominal masses encircling the 1st and 2nd portion of duodenum and multiple intrahepatic masses (Fig. 1). Enlarged lymph nodes invaded and compressed the superior mesenteric vein. A small amount of ascites was also found. Esophagogastroduodenoscopy revealed a huge protruding ulceroinfiltrative mass in the 2nd portion of duodenum. Further endoscopic examination beyond this point was impossible due to the mass (Fig. 2).
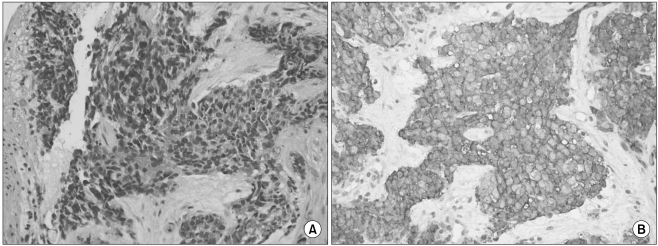
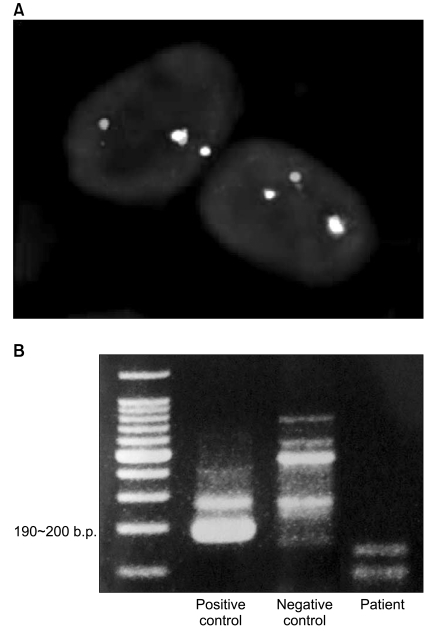
The histopathologic results from the biopsy of the duodenal mass (Fig. 3) included a poorly differentiated tumor composed of nests of small round cells with scanty cytoplasm surrounded by prominent desmoplastic stroma. The tumor cells were relatively uniform and the chromatin was evenly dispersed with inconspicuous nucleoli. Cellularity was high and nuclear molding was often observed in tumor cells. There was no observed epithelial differentiation or rosette formation. The tumor cells coexpressed a mesenchymal marker (vimentin) and a neuronal marker (neuron specific enolase). Other immunohistochemistry results were as follows: CD99 (+ in cytoplasm as well as in cell membrane), cytokeratin (-), chromogranin (-), synaptophysin (-), desmin (-), and HMB-45 (-). Histologic diagnosis was desmoplastic small round cell tumor. On fluorescence in situ hybridization (FISH) for EWSR1 (Ewing sarcoma breakpoint region 1) of EWS gene, heterozygous break apart of EWSR1 was seen in the tumor cells (Fig. 4A). The results of reverse transcriptase polymerase chain reaction (RT-PCR) were negative for EWS/Fli1 translocation, which is specific for Ewing sarcoma/PNET (Fig. 4B). The above molecular studies confirmed the diagnosis of desmoplastic small round cell tumor.
We performed a percutaneous transhepatic biliary drainage (PTBD) for proximal common bile duct obstruction. After improvement of obstructive jaundice, we inserted a stent into the obstructed bile duct through a PTBD tube and then removed the tube. Afterward, the patient was treated with palliative chemotherapy using vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide (VAC/IE). After 2 cycles of chemotherapy, the longest diameters of target tumors decreased by 14%, which was considered to be stable disease by the RECIST (Response Evaluation Criteria in Solid Tumors). The patient's general health condition recovered and she was ambulatory with an improved quality of life for several months. After 4 cycles, she experienced progressive disease and started to receive second-line chemotherapy consisting of etoposide, ifosfamide, and cisplatin (VIP). We planned debulking surgery when she had achieved a maximal response to chemotherapy. However, after 4 cycles of the second line therapy, we stopped the therapy due to poor performance status, despite the initial response of stable disease: the longest diameters of the target tumors were increased by 13%. She expired 9 months after her initial diagnosis.
DISCUSSION
DSRCT has been reported sporadically worldwide after 1st identification in 1989 by Gerald and Rosai (1). Because the tumor can have various differentiations such as epithelial, mesenchymal, or neuronal differentiation, it is difficult to diagnose DSRCT even when the immunohistochemistry results are available (3-5).
Small round cell tumor of the abdomen requires differential diagnosis for various neoplasms such as Ewing sarcoma/PNET, DSRCT, neuroendocine tumor, rhabdomyosarcoma, Wilms tumor, and lymphoma. Because the tumor did not express chromogranin and synaptophysin, we could exclude the possibility of the diagnosis of neuroendocrine tumor. Clinical and pathologic features were not compatible with rhabdomyosarcoma, Wilms tumor, or lymphoma. However, we could not completely differentiate between extraskeletal Ewing sarcoma/PNET and DSRCT using clinical features and pathologic findings in this case. The tumor expressed CD99, which is highly sensitive to Ewing sarcoma/PNET. However, many other small round cell tumors including DSRCT can express CD99 on their surfaces.
DSRCT and Ewing sarcoma/PNET have EWS gene translocations, which enable discrimination of these tumors from the other tumors. EWS gene translocations can be identified using FISH methods. While Ewing sarcoma has EWS/Fli1 translocation (t(11;22) (q24;q12)), DSRCT has EWS/WT1 translocation (t(11;22)(p13;q12)). The differential diagnosis of these diseases can be made using RT-PCR (6,7). We can exclude the possibility of Ewing sarcoma/PNET, in this case by means of RT-PCR. Several cases of DSRCT have been reported in Korea (14,15). However, there has been no report on the usefulness of molecular genetic methods in support of diagnosing DSRCT in Korea.
The prognosis of patients with DSRCT is generally poor. Kretschmar et al. reported summarized results on the prognosis of patients with DSRCT from previous reports: the median overall survival of patients was 17 months (range, 3~72) (11). Our patient also had a poor prognosis.
Chemotherapy, radiotherapy, and debulking surgery have been used to treat patients with DSRCT (8-13). However, these studies have been performed retrospectively, in relatively small populations.
There are only a few reports on the role of chemotherapy in these patients. Some reported patients who achieved radiologic complete remission after chemotherapy (8-10). Kushner et al. reported 100% response rate using a chemotherapy regimen consisting of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide in 10 patients with DSRCT (8). Farhat et al. treated 5 patients with combination chemotherapy of cisplatin, etoposide, cyclophosphamide, and anthracyclines (doxorubicine or epirubicine): 4 patients had stable disease and 1 patient had complete remission (9). Two studies on high dose chemotherapy with autologous stem cell transplantation failed to demonstrate superiority to conventional chemotherapy in terms of response rate and overall survival in patients with DSRCT (12,13). However, the populations in these studies were too small to indicate real differences in response rates between group findings Given these results, chemotherapy may be beneficial in patients with DSRCT and further comparative studies with larger population are necessary.
On the other hand, Goodman et al. reported results of whole abdominopelvic irradiation in 21 patients who had received chemotherapy followed by debulking operation. The median time to relapse of them was 19 months and the median overall survival was 32 months (10).
Our patient was treated with combination chemotherapy consisting of VAC/IE as first line palliative chemotherapy and subsequently treated with VIP second line chemotherapy. The maximum response in this case was stable disease (14% decreases) to first line chemotherapy and the time to progression after initial chemotherapy was 5 months. Although the response was not surprising, she could manage herself during palliative chemotherapy and was ambulatory during chemotherapy with improvement in abdominal pain and obstructive symptoms caused by a bulky mass effect. Reinsertion of PTBD was unnecessary during chemotherapy.
In conclusion, we report on the case of a young female patient who was diagnosed with DSRCT and treated with combination chemotherapy. Molecular genetic methods such as FISH or RT-PCR could be helpful in diagnosis of DSRCT. Chemotherapy could be an option in the treatment of patients with DSRCT.
 XML Download
XML Download