

 PDF
PDF Citation
Citation Print
Print


Introduction
Conventional chemotherapeutic drugs are distributed non-specifically in the body where they affect both cancerous and healthy cells, resulting in dose-related side effects and inadequate drug concentrations reaching the tumor. Non-specific drug delivery leads to significant complications that represent a serious obstacle to effective anticancer therapy. In addition, the occurrence of resistance phenomena reduces the efficacy of cancer treatment. To overcome the lack of specificity of conventional chemotherapeutic drugs, several ligand-targeted therapeutic strategies, including immunotoxins, radioimmunotherapeutics, and drug immunoconjugates, are being developed. Although these conjugated agents have shown promising efficacy compared with conventional chemotherapy drugs, limitations in their delivery efficiency still remain.
Recent progress in cancer nanotechnology raises exciting opportunities for specific drug delivery. Nanoparticles, particularly in the size range from 10 nm to 100 nm, are emerging as a class of therapeutics for cancer treatment. Nanoparticles can be composed of several functional molecules simultaneously, such as small molecule drugs, peptides, proteins, and nucleic acids. By using both passive and active targeting strategies, nanoparticles can increase the intracellular concentration of drugs in cancer cells while minimizing toxicity in normal cells; thereby enhancing anticancer effects and reducing systemic toxicity simultaneously, when compared with the therapeutic entities they contain. Furthermore, nanoparticles offer the potential to overcome drug resistance, since nanoparticles can bypass the P-glycoprotein efflux pump, one of the main drug resistance mechanisms, leading to greater intracellular accumulation.
The purpose of this review article is to summarize the results of the use of therapeutic nanoparticles in the clinic and discuss the opportunities and challenges faced by therapeutic nanoparticles. Thus, the first part will emphasize the key properties of therapeutic nanoparticles and how these properties affect the efficiency and specificity of nanoparticles as a drug delivery system. Next, we will summarize current clinical uses of the first-generation therapeutic nanoparticles and the advances in new generation of therapeutic nanoparticles currently under preclinical and clinical investigation. Finally, we will discuss how nanoparticles will be developed to improve their therapeutic efficacy and function for future cancer treatment.
Nanoparticles for Tumor Targeting and Delivery
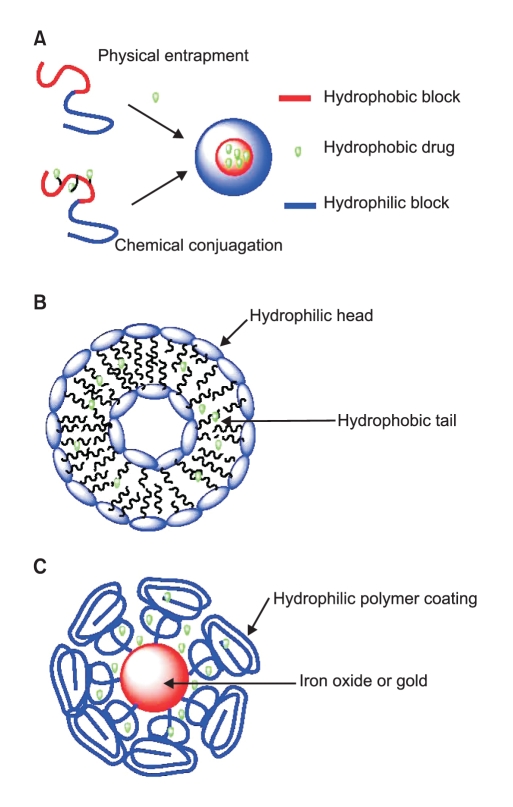
Nanoparticles used for anticancer drug delivery can be made from a variety of materials, including polymers, dendrimers, liposomes, viruses, carbon nanotubes, and metals such as iron oxide and gold (Fig. 1). So far, almost all the nanoparticle delivery systems which have been approved by the FDA or are currently in clinic trials are based on polymers or liposomes (1).
1. Polymeric nanoparticles
Generally, polymers that are used for preparation of nanoparticles fall into two major categories: natural polymers and synthetic polymers. A number of natural polymers such as heparin, dextran, albumin, gelatine, alginate, collagen, and chitosan have been intensively investigated. Synthetic polymers including polyethylene glycol (PEG), polyglutamic acid (PGA), polylactic acid (PLA), polycarprolactone (PCL) and N-(2-hydroxypropyl)-methacrylamide copolymer (HPMA) have been exploited as well. General requirements for those polymers are biocompatibility, biodegradability, and their capacity to be functionalized (2).
The formation of polymeric nanoparticles has been summarized by several review articles (3,4). In most cases, the polymeric nanoparticle consists of two parts, a hydrophobic core which serves as the container for anticancer agents and a hydrophilic shell which stabilizes the nanoparticle in aqueous environments.
The drug can be loaded into polymeric nanoparticles through two methods: by physical entrapment or by chemical conjugation. A hydrophobic interaction between the core of the polymeric nanoparticle and the drug molecule allow the drug to be entrapped in the nanoparticle core. For instance, deoxychilic acid-modified heparin can self-assemble into 100~200 nm nanoparticles (5) and its hydrophobic core can be used to entrap 4~12% of the total weight of doxorubicin (6) When the drug molecule is covalently conjugated onto the polymer, the chemical properties of the linker between the drug and polymer are critical. If the linker is too stable, drug release may be delayed, while if the linker is too unstable, drug may be released before the nanoparticle reaches the tumor. Therefore, a proper linker is very important to the drug-polymer conjugate. A variety of pH-sensitive linkers have been developed such as hydrozone and cis-aconityl (7,8). These chemical bonds are stable in the blood circulation system (pH=7), but quickly decompose and release drug molecules inside the tumor where pH values typically drop below 5.5. Disulfide bonds are also very attractive because they can be cleaved by glutathione. The intracellular level of glutathione is much higher than its extracellular level, therefore, the disulfide linker is relatively stable while in blood circulation and becomes unstable and releases the drug molecules once it is internalized by cells (9,10).
It is important to note that dendrimers, synthetic superma cromolecules with highly branched repeated three dimensional structures, have emerged as important materials for biological application due to their unique features such as the precise control of size and shape, uncommon physical properties, controlled degradation, and the ability to place numerous functional groups on their periphery and/or core (11,12). There are more than 50 different types of dendrimers (13). Among them, polyamidoamine and poly (propylenemine) have been commercialized and used extensively as biomaterials in gene and drug delivery (14,15) and for nanoparticle encapsulation (16,17) in imaging (18,19).
2. Liposomal nanoparticles
Liposomes are self-assembling spherical particles with a membrane composed of phospholipid bilayers. The size of liposomes can range from 25 nm to 10 µm depending on the preparation method. They have been studied as candidates for drug delivery for the last 50 years since being first discovered by Bangham (20). The synthesis of liposomal nanoparticles has been reviewed by Bellare, et al (21). Drug delivery systems based on unmodified liposomes are limited by their short blood circulation time. This is mainly due to the fast clearance of liposomes by macrophages of the reticuloendothelial system (RES) (22). The second generation of polymer-coated liposomes can dramatically increase blood circulation times from several minutes up to 3 days.
3. Gold and iron oxide nanoparticles
Recently, several novel nanotechnology concepts have been applied to the development of a new generation of anti-cancer drug delivery systems. Gold nanoparticles can be synthesized through the reduction of HAlCl4 with a very narrow polydispersity. Several gold nanoparticle anticancer drug delivery systems have been reported and showed good in vitro results (23).Although it seems that gold is inert under physiological environments, the long term toxicity of gold nanoparticles remains an unanswered question. One attractive property of gold nanoparticles is that gold concentrations are naturally low in animal bodies, allowing the convenient use of this nanoparticle model for in vivo pharmacokinetic and biodistribution studies (24).
It is worth mentioning that a gold nanorod formulation is showing very promising potential as a photothermal therapy agent. Gold nanorod can generate heat when it is radiated by a near infra-red (IR) laser (wavelengt > 650 nm). At this range, the laser is relatively safe to the tissue and organs. Once the gold nanorod has accumulated inside the tumor through passive/active targeting, it can be heated locally up to 43℃ (25) by radiation with a near IR laser to destroy the tumor without causing damage to surrounding healthy tissues (27).
Iron oxide nanoparticles have been clinically used as imaging agents for MRI. Recently a number of research groups have investigated them as drug carriers while retaining their imaging functions (26-28). One unique advantage of iron oxide nanoparticle delivery systems is that they can be delivered in a targeted manner to a desired region by applying an external magnetic field.
Properties of Nanoparticles
A suitable nanoparticle size is very important for efficient drug delivery. Generally, 10~100 nm is considered the optimal size for nanoparticle drug carriers. If the particle size is less than 10 nm, the nanoparticles will be quickly eliminated by renal clearance (threshhold < 6 nm). At sizes greater than 100 nm, the chance of the particle being captured by the RES will dramatically increase (29).
A proper surface coating is essential to the stability and circulation time of nanoparticle delivery systems. For instance, a sodium citrate-stabilized gold particle aggregates in PBS within several minutes. But once coated with thiol-terminated polyethylene glycol (PEG) polymer, this nanoparticle is stable not only in PBS but also under low or high pH conditions (32). Generally, a neutral-charged nanoparticle can achieve a long circulation time and reduce the chance of nanoparticle capture by the immune system.
Targeted Delivery of Therapeutic Nanoparticles
1. Passive targeting
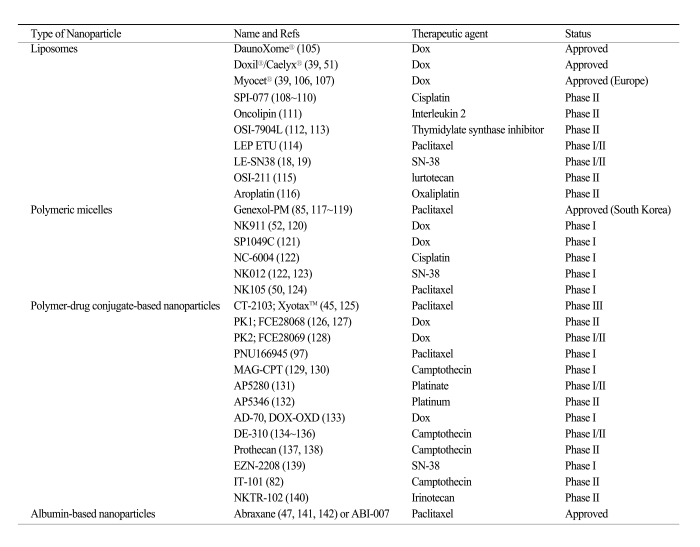
Table 1 lists nanoparticles that have been used in the clinic and utilize passive targeting to achieve their selective delivery to tumors. Passive targeting takes advantage of the inherent size of nanoparticles and the unique properties of tumor vasculature (30-33). As tumors grow and begin to outstrip the available supply of oxygen and nutrients, they release cytokines and other signaling molecules that recruit new blood vessels to the tumor in a process called angiogenesis. Unlike the tight blood vessels in normal tissues, angiogenic blood vessels in tumor tissues have gaps as large as 600 to 800 nm between adjacent endothelial cells (34,35). This defective vascular architecture coupled with poor lymphatic drainage induces an enhanced permeability and retention effect (EPR) (36,37), Through these gaps, nanoparticles can selectively accumulate into the tumor interstitium (38) (Fig. 2).
In general, the accumulation of nanoparticles in tumor tissues is dependent on several factors including the size, surface characteristics, and circulation half-life of the nanoparticles and the degree of angiogenesis of the tumor (39). It is speculated that nanoparticles with a size between 10 and 100 nm will be optimal for tumor accumulation. For example, smaller polymeric micelles (20 nm) have been shown to accumulate more readily in tumors than larger liposomes (100 nm) (40,41). Proper surface characteristics and longer circulation times of nanoparticles can also improve tumor uptake, as described earlier. The unmodified phospholipid surface of liposomes can attract plasma proteins and thus recognition by the mononuclear phagocytic system (MPS), resulting in their rapid clearance from the circulation. This property impedes the distribution of liposome-associated drugs to solid tumors. Surface-modified (stealth) liposomes have solved the problem of fast clearance from the circulation, yielding liposomes with a significantly increased half-life in the blood (42,43). Dramatically reduced clearance rates have also been obtained with other nanoparticles such as Abraxane (44), Xyotax (45) and IT-101 (46). Tumor vascularization also affects nanoparticle accumulation; usually nanoparticles accumulate poorly in poorly vascularized tumors, small pre-angiogenic tumors, or large necrotic tumors.
As drug delivery systems, nanoparticles have shown an ability to improve pharmacokinetics, pharmacodynamics, efficacy, and to reduce the toxicity of associated drugs (40). For example, Abraxane (ABI-007), an albumin-bound nanoparticle of paclitaxel (Taxol) which has been approved for the treatment of metastatic breast cancer, showed significant greater efficacy than free paclitaxel in a phase III clinic trial (47). Despite the increased dose of paclitaxel in the Abraxane group, the incidence of grade 4 neutropaenia was significantly lower than in patients treated with free paclitaxel. Pharmacokinetic studies also showed that paclitaxel clearance and the volume of distribution were higher for Abraxane than for paclitaxel: Clearance was 13 litres per hour per m2 for Abraxane versus 14.76 litres per hour per m2 for paclitaxel (p=0.048), and distribution was 663.8 litres per m2 for Abraxane versus 433.4 litres per m2 for paclitaxel (p=0.04) (44). Similar to Abraxane, NK105, a micellar nanoparticle formulation of paclitaxel also showed improved pharmacokinetics, pharmacodynamics, efficacy, and reduced toxicity as compared with free paclitaxel in a preclinical study and a phase I trial (48,49). The plasma area under the curve (AUC) value was approximately 90-fold greater for NK105 than for free paclitaxel and the tumor AUC value was 25-fold higher for NK105 than for free paclitaxel in an animal model (48). In patients, the plasma AUC of NK105 at 180 mg/m2 was approximately 30-fold greater than that of the conventional formulation of paclitaxel (49). NK105 showed significantly more potent antitumor activity in a human colorectal cancer cell line HT-29 xenograft than free paclitaxel, due to enhanced accumulation of the drug in the tumor (48). The phase I trial showed that NK105 was well tolerated and effective in patients with pancreatic cancer (50). These differences in pharmacokinetic properties may contribute to the increased drug accumulation inside the tumor observed with nanoparticles compared with the corresponding free drugs. Other nanoparticles currently used in the clinic or undergoing clinic trials also showed an improved pharmacokinetic profile compared with the respective free drugs, such as Doxil, a PEG-liposome loaded with doxorubicin (DOX) (51), SP1049C, a pluronic micelle loaded with DOX (40), NK911, a PEG-Asp micelle loaded with DOX (52), and Xyotax, a polyglutamic acid nanoparticle carrying paclitaxel (45).
2. Active targeting
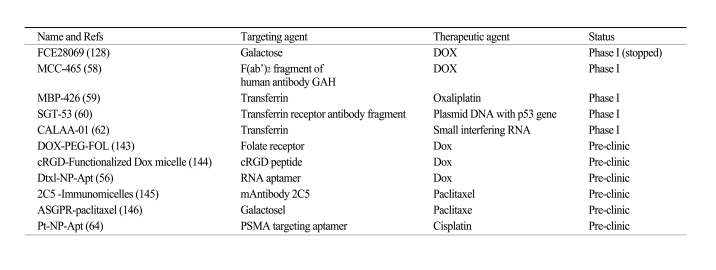
The nanoparticles listed in Table 1 that have been used in the clinic so far mostly utilize the EPR effect of tumors and the tumor microenvironment to promote their selective delivery to tumors. However, certain limitations of non-targeted nanoparticles as a drug delivery system still remain. For example, in the case of the EPR effect, although poor lymphatic drainage helps the extravasated drugs to be enriched in the tumor interstitium, it also induces drug outflow from the cells as a result of higher osmotic pressure in the interstitium, which eventually leads to drug redistribution in some portions of the cancer tissue (53). Most importantly, accumulation merely within the tumor microenvironment by the EPR effect may not always correlate with therapeutic efficacy since internalization into the tumor cells is required for most anticancer drugs to exert their biological functions. To overcome these limitations, a rational approach is to incorporate a targeting moiety on the nanoparticle surface. The targeting moiety is expected to bind a tumor-associated antigen or receptor and facilitate the delivery of nanoparticles to the intracellular site of drug action, enabling a greater therapeutic effect (Fig. 3). Recent preclinical studies have shown that targeted nanoparticles have better antitumor activity compared with non-targeted nanoparticles (54-57). Although targeted nanoparticles may not always mediate an increase in tumor drug accumulation when compared with non-targeted nanoparticles, targeted nanoparticles show greater intracellular drug delivery to cancer cells than non-targeted nanoparticles, resulting in dramatically increased antitumor efficacy (54-56). These findings suggest that the primary role of the targeting ligands is to enhance cellular uptake into cancer cells and to minimize cellular uptake in normal cells.
Although current studies have shown that the use of targeted nanoparticles as a drug delivery system is a promising strategy to treat human cancers, it is still in its early stage of development. Clinical data using targeted nanoparticles are limited since most targeted nanoparticles have not yet reached the clinic. Only a few targeted nanoparticles are currently under clinical investigation. One is MCC-465, which is an immunoliposome-encapsulated doxorubicin (Dox). The liposome is tagged with PEG and the F(ab')2 fragment of the human monoclonal antibody GAH, which recognizes a cell surface molecule on various types of cancer cells (58).Phase I studies have indicated that the PK parameters of MCC-465 differ from those of free Dox, but were very similar to those of Doxil (non-targeted liposome-encapsulated Dox) in humans. In terms of skin toxicity, the patients who received MCC-465 did not experience any severe skin toxicity such as palmar-plantar erythrodysesthesia (PPE) or mucositis, unlike the patients who received Doxil (58). Besides MCC-465, other examples of targeted therapeutic nanoparticles include MBP-426 which contains the cytotoxic platinum-based drug oxaliplatin in a liposome (59), SGT-53, a liposome containing a plasmid coding for the tumor suppressor p53 (60), and CALAA-01, a polymer-siRNA conjugate (61,62) (Table 2) . These nanoparticles all target the transferrin receptor which is upregulated in many types of cancer (63).
3. Selection of target receptor and ligand
Selection of the target receptor or antigen on cancer cells is crucial for the optimal design of targeted nanoparticles. In general, cell-surface antigens and receptors should have several properties that render them particularly suitable as tumor-specific targets. First, they should be abundantly and uniquely expressed on tumor cells, but negligibly or less expressed on normal cells. Second, they should have a high density on tumor cells.
A targeting ligand should selectively and successfully transport nanoparticles into targeted cancer cells. It is believed that internalization of nanoparticles after binding to targeted tumor cells is necessary for good therapeutic responses, so whether the targeted nanoparticles can be internalized is an important issue in the selection of proper targeting ligand. Use of a ligand that can not trigger the internalization process may result in drug release outside the cell and its redistribution to the surrounding normal tissues.
Reduction of Multidrug Resistance
Drug resistance remains one of the major challenges in cancer therapy. A number of mechanisms for drug resistance have been described. Drug resistance can be caused by physiological barriers (non-cellular based mechanisms), or alterations in the biology and biochemistry of cancer cells (cellular mechanisms). Non-cellular drug resistance can be caused by poorly vascularized tumor regions and/or physiological barriers that greatly reduce drug access to the tumor tissues, thus protecting cancerous cells from drug-induced cytotoxicity. Cellular drug resistance can be due to overexpressed drug export pumps, such as P-glycoprotein (p-gp) and other drug-resistance proteins, increased DNA repair capacity, and reduced apoptosis regulation.
Among these mechanisms, the roles of the drug efflux pumps are the most extensively investigated. P-glycoprotein (p-gp), a product of the MDR1 gene, is a 170-kD transmembrane glycoprotein that functions as an efflux pump to remove drug from cells. Several specific p-gp inhibitors have been investigated to overcome drug resistance. Although in preclinical studies, some of these p-gp inhibitors have shown the restoration of cancer cell sensitivity to anticancer drugs, the clinical trials results have been disappointing (77,78).
Alternative strategies for overcoming drug resistance have been studied. Newly developed drug delivery systems, including nanoparticle, allow selective drug accumulation in tumor tissues, tumor cells, or even compartments of tumor cells. Because with the aid of a targeting moiety nanoparticles enter cells through endocytosis, it is expected that they can bypass the p-gp efflux pump, leading to their greater intracellular accumulation (Fig. 3). Many nanoparticles have been used to overcome or minimize drug resistance in preclinical studies and the results are very promising. For example, doxorubicin (DOX)-loaded poly (alkyl cyanoacrylate) nanoparticles (79), PACA nanoparticle (80,81), and IT-101 (a nanoparticulate conjugate of 20 (S)-camptothecin) (82) have shown the ability to overcome drug resistance in the tested models. And most importantly, the ability of targeted nanoparticles to overcome drug resistance has been confirmed in humans. It has been demonstrated in clinical studies that liposomal doxorubicin is able to overcome drug resistance in AIDS-related Kaposi's sarcoma (83,84). Also, clinical trials showed positive results using nanoparticles in patients who had previously failed chemotherapy (83,85,86). Ligand-targeted strategies, especially those using receptor-targeting ligands, have also been applied to overcome drug resistance since these ligands are internalized via receptor-mediated endocytosis, bypassing the plasma membrane where p-gp primarily acts. As an example, folate receptor-targeted pH-sensitive polymeric micelles containing DOX (87) and transferrin-conjugated paclitaxel nanoparticles exhibited greater cytotoxicity than the respective free drugs in a drug-resistant model (88). As illustrated, using nanoparticles as a drug delivery system may be able to overcome certain kinds of cancer drug resistance.
Potential Toxicity of Nanoparticles
An important consideration in nanoparticle development is the biological behavior of carrier constituents and their potential toxicity, especially during chronic administration. Many candidate polymers have been defined with particular toxicities, such as hematotoxicity, complementactivation, carcinogenicity, teratogenicity, and immunogenicity (89,90), indicating the importance of choosing safe polymers for the design of nanoparticles. In addition, the biological properties of polymers are molecular weight-dependent and can be changed once the respective conjugates are prepared. Therefore, careful characterization of the potential toxicity of both the polymer and the final nanoparticle is critically important. For non-biodegradable polymers, potential toxicity is concerning when the polymer molecular weight is greater than the renal threshold. Increased understanding of the potentially deleterious properties of polymers leads to the design of new and safer polymeric nanoparticles.
Currently, most nanoparticles use nontoxic and biodegradable ingredients, so toxicities associated with the carrier molecules per se tend to be mild. However, particular nanoparticles cause increased accumulation of drugs in MPS cells in the liver, spleen, and bone marrow, with the possibility of increased toxicities to these organs. Among these organs, the liver has been identified in many studies as the primary organ responsible for reticuloendothelial capture of nanoparticles, often due to phagocytosis by Kupffer cells (91,92). Hepatic uptake has been shown to be a main mechanism of hepatic clearance from the blood circulation following the intravenous injection of nanoparticles. In addition to hepatic accumulation, some nanoparticles have been reported to cause liver injury (decreased function and hepatic morphology changes) (93,94). For example, intravenous administration of cationic PAMAM dendrimers caused liver injury when administered intravenously to mice (95). Hepatotoxicity has also been observed in mice treated orally with nano-zinc particles (96). Also there are safety concerns with particular nanoparticles that are able to cross the blood brain barrier. Lessons have been learned from many of the early clinical studies. For example, due to neurotoxicity, a clinical trial testing an HPMA-conjugated paclitaxel was terminated (97). The failure of MAG-camptothecin due to cumulative bladder toxicity in phase I was also reported (98).
Attempts are being made to decrease the uptake of nanoparticles by MPS cells and to increase their accumulation in the active site, through polymer or nanoparticle surface modifications, and/or incorporating targeting ligands (54,99,100). With more rational design, many nanoparticles have shown an improved safety profile and enhanced antitumor efficacy compared with free drugs in preclinical and clinical studies (100-104). For example, Doxil (PEG-liposome loaded with doxorubicin) showed a reduction in cardiotoxicity over that of doxorubicin in a clinical study (103,104). Abraxane (albumin nanoparticle loaded with paclitaxel) showed a greater therapeutic outcome compared with free paclitaxel and, taking advantage of the water solubility of the nanoparticle, successfully eliminated the side effects associated with the toxic vehicle Cremophor EL (47).
Implications and Future Directions
Nanoparticles provide opportunities for designing and tuning properties that are not possible with other types of therapeutic drugs, and have shown a bright future as a new generation of cancer therapeutics. Furthermore, the development of multifunctional nanoparticles may eventually render nanoparticles able to detect and kill cancer cells simultaneously. Although there are certain critical questions and many challenges remaining for the clinical development of nanoparticles, as more clinical data are available, further understanding in nanotechnology will certainly lead to the more rational design of optimized nanoparticles with improved selectivity, efficacy, and safety.


 XML Download
XML Download