

 PDF
PDF Citation
Citation Print
Print


Introduction
Although EES is extremely rare, it responds relatively well to a combination of surgical resection, chemotherapy and radiation therapy. Thus, an aggressive treatment protocol is needed. As there have been few reports on EES, clinicians should devise a more comprehensive and accurate pathological diagnostic method and a more systematic clinical evaluation method for the family of small round cell neoplasms of bone and soft tissue. We have reported two cases of EES that were successfully treated with combined multimodal treatment.
Case Reports
Case 1
A 23-year-old man presented at our hospital with a one-month history of a palpable mass located at the sixth intercostal space on the midmediastinal line of the right anterior chest. At the time of presentation, vital signs were normal and there were no other symptoms or signs such as fever. Routine blood tests and chemical tests showed no abnormal findings. The patient reported that the mass was first noticed one month earlier and had become slightly enlarged, but the patient did not complain of chest pain.
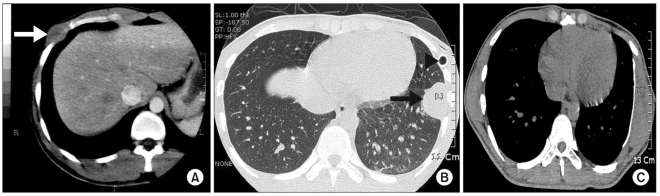
There was no remarkable history of medical illness, drinking or smoking. On a physical examination, a soft, non-tender and non-nodular mass that measured approximately 4 cm in diameter was palpated in the sixth intercostal space on the right anterior chest. Chest X-rays demonstrated the presence of a mass shadow without any other abnormalities. A chest computed tomography (CT) examination showed the presence of a 3.5×2.3 cm round, low-density shadow in the sixth right intercostal space that was enhanced peripherally without invasion to the lung (Fig. 1). There were no abnormal lesions in the lung or enlargement of regional lymph nodes. Chest ultrasonography demonstrated the presence of a 3.4 cm round mass without increased blood flow.
A bone scan showed no abnormal findings, and other preoperative tests were normal. A histological examination of a biopsy specimen showed the presence of a small round cell malignancy. Under general anesthesia, an incision was made in the right sixth intercostal space in the left lateral decubitus position. The mass did not invade the lung and adjacent tissue. The sixth and the seventh ribs along with the mass were resected with resection margins of 4 cm.
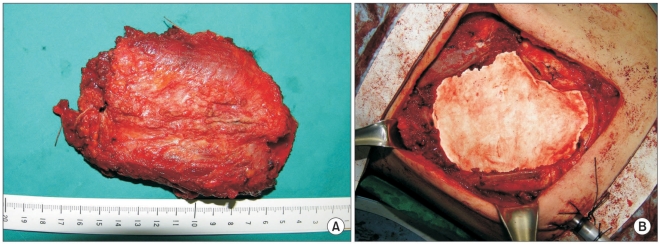
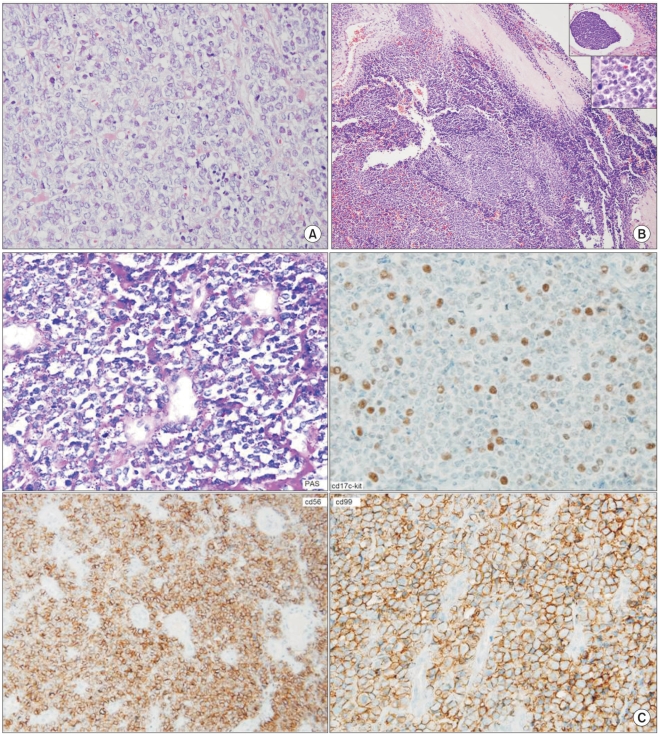
A frozen section examination was negative for malignancy as seen at the resection margin and bony structure. The surgical defect in the chest wall that measured 9×13 cm was repaired with a Gore-Tex patch (Fig. 2). A macroscopic examination of the resected mass showed the presence of a clear resection margin and no invasion or destruction of the rib. A diagnosis of extraskeletal Ewing's sarcoma (EES) without invasion or destruction was made based on the histological examination of the resected tissue. The mass measured 5.0×3.0×2.5 cm and did not invade the adjacent tissue. Periodic acid-Schiff (PAS) staining was positive in the cytoplasm, and immunohistochemical staining was positive for CD99, CD56 and vimentim but was negative for CD45, cytokeratin, EMA, S100 and desmin (Fig. 3).
The chest tube was removed five days after surgery and the patient was discharged from the hospital on the tenth postoperative day. The patient consulted an oncologist regarding chemotheraphy and consulted a radiation oncologist regarding the use of radiation therapy. The patient began to receive anticancer chemotherapy with the VAC regimen (2 mg vincristine on day 1, 45 mg/m2 adriamycin on days 1 and 2 for 15 minutes, 900 mg/m2 cyclophosphamide for 30 minutes with 350 mg/m2 mesna per 4 hours on days 1 and 2) during the tenth postoperative week. The IE regimen (1,000 mg/m2 ifosfamide for 60 minutes with 350 mg/m2 mesna per 4 hours on days 1, 2, 3, 4 and 5 and 100 mg/m2 etoposide for 90 minutes on days 1, 2, 3, 4 and 5) was administered during the 13th postoperative week. The VAC regimen was administered during the 16th and 22nd postoperative weeks, and the IE regimen was administered during the 19th, 25th, 28th, 31st, 37th, 43rd, 49th, 55th, 58th and 61st weeks. The VaC regimen (2 mg vincristine on day 1, 35 mg/m2 adriamycin on days 1 and 2 for 15 minutes, 900 mg/m2 cyclophosphamide for 30 minutes with 350 mg/m2 mesna per 4 hours on days 1 and 2) was administered during the 34th, 40th, 46th and 52nd postoperative weeks. The patient underwent follow-up without any local recurrence or distant metastasis for 30 months after surgery.
Case 2
An 18-year-old male patient presented at the emergency department with left chest pain that had developed on the day of presentation. The vital signs were normal, but respiratory sound decreased on the left side. Chest X-rays demonstrated the presence of a spontaneous pneumothorax on the left side. A thoracostomy was performed. Routine blood tests and chemical tests revealed no abnormal findings, and there were no specific past histories of diseases, alcohol consumption and smoking. Chest X-rays obtained after chest tube insertion demonstrated the presence of an expanded lung on the left side and a mass shadow in the left inferior lobe. Chest CT scans showed multiple pneumocystic changes in the left upper lobe and a 5.0×2.5 cm round low-density shadow. The mass was enhanced peripherally, but there was no invasion of the lung. Chest ultrasonography demonstrated the presence of a 5 cm round mass without an increase in blood flow and any invasion of the adjacent tissue. Bone scan findings and other preoperative laboratory tests were normal.
There was no evidence of the presence of a distant metastasis. A histological examination of a biopsy specimen demonstrated the presence of a small round cell malignancy. Under general anesthesia, surgery was performed in the right lateral decubitus position. Wedge resection of the left upper lobe was performed using a thoracoscope that was inserted through the fourth intercostal space and the wound where the chest tube was placed for the pneumothorax lesion. A lateral incision was made in the seventh intercostal space for removal of the mass. There was no invasion of the lung and adjacent tissue. The sixth, seventh and the eighth ribs along with the mass were resected with resection margins of 4 cm. A frozen section examination was negative for malignancy at the resection margin and bony structure. The surgical defect that measured 10×15 cm was repaired with a 2-mm Gore-Tex patch (Fig. 2).
A macroscopic examination of the resected tissue showed the presence of a clear resection margin and no invasion or destruction of the rib. A microscopic examination confirmed the dignosis of EES without invasion or destruction of the rib. The mass measured 5.1×2.3×2.1 cm and did not invade the adjacent tissue. PAS staining was negative in the cytoplasm. Immunohistochemical staining was positive for CD99, CD56 and vimentin but was negative for CD45, cytokeratin, EMA, S100 and desmin (Fig. 3).
The chest tube was removed on the sixth postoperative day, and the patient was discharged from the hospital on the 11th postoperative day. Chemotherapy was performed in the same manner as for case 1 and radiation therapy commenced from week 40 to deliver 54 Gy with 1.8 Gy per fraction to the chest wall tumor bed. There was no local recurrence or a distant metastasis detected a 22-month follow-up examination (Fig. 1).
Discussion
Since J. Ewing first described Ewing's sarcoma in 1921, it has been reported as the second most common malignant bone tumor. Ewing's sarcoma usually develops in the bone and invades the adjacent soft tissue in 90% of cases. Ewing's sarcoma rarely develops in extraskeletal regions. ESS may develop in the soft tissue of the trunk or extremities and may cause pathological changes in the cortex of the adjacent bone. Ewing's sarcoma is more prevalent in males than females, whereas there is no significant difference in the prevalence of ESS between the sexes.
ESS occurs more frequently in adolescents and young adults. ESS involves a wider area as compared to Ewing's sarcoma. ESS commonly occurs in the chest wall and around the spine, but it has been reported that ESS sometimes occurs in the pelvis, retroperitoneal cavity, diaphragm and duodenum (1).
ESS should be differentiated from an embryonal rhabdomyosarcoma, lymphoma and neuroblastoma. ESS and Ewing's sarcoma have similarities for histological findings, immunohistochemical staining results and molecular biological characteristics. EES is histologically defined as having small homogenous, round or oval cells containing solid material separated by fibrous septation. In addition, since the cytoplasm of EES cells contains glycogen, EES cells produce vacuoles and are not easily stained (2). ESS should also be distinguished from a primitive neuroectodormal tumor (PNET), neuroblastoma and embryonic rhabdomyosarcoma using clinical features, immunohistochemical staining and cytogenetic analysis. In particular, some pathologists regard ESS as a completely different disease entity from PNET, whereas other pathologists consider the two diseases as belonging to the same disease group. Although both ESS and PNET show expression of HBA-7 and the t11:22 translocation, only ESS contains PAS-stained glycogen in the cytoplasm (3).
A macroscopic examination shows the filigree pattern in Ewing's sarcoma, with the presence of homogeneous solid segmented plasma cells, densely distributed blood vessels and degenerated/necrotized ghost cells. Immunohistochemical staining is useful to diagnose Ewing's sarcoma. Expression of the MIC2 gene is pathognomonic for Ewing's sarcoma, but expression of MIC2 occurs in some cases of T-lymphoblastic lymphoma, poorly differentiated synovial sarcoma, small cell osteosarcoma and rhabdomyosarcoma. It has been reported that in patients with Ewing's sarcoma, immunohistochemical staining is positive for CC99 and CC56 but is negative for S-100, demin, factor 8 cytokeratin and neurofilament as well as for T-cells, B-cells, LCA, CD 43 and CD68. Ewing's sarcoma can be definitively diagnosed by detection of the EWS gene and rearrangement of ETS-related cancer genes. The identification of the Ewing's sarcoma breakpoint region (EWSR1) is helpful to diagnose Ewing's sarcoma (4).
The most effective treatment for EES is surgical resection along with postoperative chemotherapy and the use of high-dose radiation therapy. Complete removal of the tumor is essential whenever feasible. Treatment outcome after chemotherapy and radiation therapy has not yet been determined. It has been demonstrated that preoperative chemotherapy with the use of a combination of vincristine, doxorubicine, cyclophosphamide and etoposide allows for conservative surgical resection with subsequent improvement of the postoperative condition (5). Raney et al. (6) have reported that the 10-year survival rate of Ewing's sarcoma is 61% to 77% after the use of combined chemoradiotherapy. If complete resection of Ewing's sarcoma is feasible, the survival rate increases. In addition, Desai et al. (7) have reported excision of the tumor should be as radical as possible to minimize the tumor mass and to increase the effectiveness of adjuvant therapy. Rosen et al. (8) have introduced the use of multidrug chemotherapy, which is more effective than single-drug chemotherapy for the treatment of Ewing's sarcoma. The drugs used in the study by Desai et al. (7) were etoposide, vincristine, doxorubicin, cyclophosphamide, iodophosphamide and dactinomycin.
However, as the prevalence of EES is relatively low, there are no accurate reports on long-term results. Bone marrow transplantation has been recently performed on patients with EES (6).
Major prognostic factors include the absence or presence of a metastasis, tumor size, the extent of necrosis, the initial response to chemotherapy and the presence of EWS/FL 11 fusion transcripts. Ahmad et al. (5) have reported that for the relationship between treatment outcome and patient age at the time of diagnosis, treatment outcome and the 5-year survival rate and disease-free survival were significantly higher in patients <16 years as compared to patients ≥16 years (77% versus 46%, p=0.045 for the 5-year survival rate; 77% versus 27%, p=0.005 for disease-free survival).
Although EES is extremely rare, it responds relatively well to a combination of surgical resection, chemotherapy and radiation therapy. Thus, an aggressive treatment protocol is needed. As there have been few reports on EES, clinicians should devise a more comprehensive and accurate pathological diagnostic method and a more systematic clinical evaluation method for the family of small round cell neoplasms of bone and soft tissue. We have reported two cases of EES that were successfully treated with combined multimodal treatment.
 XML Download
XML Download