

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Ataxia-telangiectasia mutated (ATM) kinase is a serine-threonine kinase that is related to phosphotidylinositol-3 kinase and is activated by DNA damage (1). ATM activation leads to diverse DNA damage responses, including activation of cell cycle checkpoints, DNA repair, and apoptosis. When the atm gene is mutated, these signaling networks are impaired, and cells do not respond appropriately in order to effectively minimize damage. Normal signaling of these pathways occurs through the ATM-mediated phosphorylation of a number of proteins, including BRCA1/Rad51/BRCA2, Nbs1/Mre11/Rad50, Chk2, FANCD, and p53/MDM2 (2-5). Phosphorylation of these proteins appears to be important for the optimal regulation of their activities during DNA damage responses. For example, ATM-mediated phosphorylation of the transcriptional activator, p53, is required for its function in cell cycle arrest and apoptosis (6-8).
Histone acetylation/deacetylation, which is mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively, plays an important role in the modulation of gene transcription (9-11). In addition to histones, the HATs and HDACs can modify the expressions of various proteins including p53, AML, and GATA (12-13). HDAC inhibitors have been reported to enhance the transcriptional activity of p53 in an acetylation-dependent manner (14-15). The well-known p53 target gene, p21WAF1, which is involved in inhibition of cell cycle progression at G1-phase, is upregulated by histone deacetylase inhibitors (1, 15-16). Furthermore, exposure of cells to chromatin-modifying drugs (including TSA) induces rapid, diffuse phosphorylation of the ATM protein, which suggests that ATM activation may result from changes in chromatin structure (17). In addition, significant decondensation of nuclear chromatin has been associated with A-T (18), and ATM has been shown to phosphorylate HDAC1 (19). Taken together, these observations suggest that histone acetylation may regulate DNA damage responses in many ways. However, molecular mechanisms in which histone deacetylation/acetylation interplays with the activation of the ATM-mediated DNA damage signal pathway via phosphorylation of many target proteins have not yet been investigated.
In this study, we sought to examine the activation of the ATM-mediated DNA damage signal pathway in response to HDAC inhibition. To this end, we examined the phosphorylation of ATM-target proteins, including ATM, H2AX, and Chk2, in isogenic A-T (ATM-), and control cells (ATM+) following treatment with TSA. We also investigated whether HDAC inhibition induces the formation of H2AXγ foci. Taken together, our findings indicate that the activation of ATM can be induced by TSA. Further, our findings suggest that aberrant chromatin remodeling may function as a DNA damage stress.
MATERIALS AND METHODS
1) Cell culture and transfection
AT22IJE pEBS7 (ATM-) and AT22IJE pEBS7-YZ5 (ATM+) cells (gifts from Y. Shiloh) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 µg/ml hygromycin B and 15% fetal bovine serum (FBS; Hyclone, Logan, UT) in humid incubators at 5% CO2 and 37℃. The human HeLa and U2OS cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. 293T cells were transfected with Flag-tagged wild-type (Flag-ATM-WT) and kinase-dead ATM (Flag-ATM-KD) expression vectors (20) using the Effectene transfection kit (Qiagen). One day after transfection, cells were incubated for another 24 h with TSA (Sigma Chemical Co.) harvesting.
2) Immunofluorescence confocal analysis
HeLa cells were exposed to γ-irradiation at 1 Gy or 12 Gy, and the cells were harvested at the indicated time points: 1 h, 4 h, 8 h, 12 h, 24 h, and 36 h, respectively. HeLa and U2OS cells were incubated for 1 h, 8 h, 12 h, 24 h, and 36 h with 0.33, 1, or 10 µM TSA. ATM- and ATM+ cells were treated with 0.33, 1, or 10 µM TSA for 24 h. These cells were then fixed with 3.7% formaldehyde for 10 min and incubated in the blocking solution (1% chicken egg albumin in 1×PBS containing 0.1% Tween 20) at room temperature for 30 min. Cells were incubated with anti-γH2AX (Upstate), anti-phospho-Ser1981 ATM (Rockland), and anti-phospho-Thr68 Chk2 (Cell Signaling) for 1 h, and were then incubated with the fluorescent antibody (Alexa Fluor488, Molecular Probes) in the dark for 1 h. DAPI (Sigma) staining was subsequently carried out. Cells were analyzed under a confocal microscope (ZEISS).
3) Immunoblotting
293T cells were exposed to γ-irradiation or treated with TSA, respectively. The cells were lysed in lysis buffer containing 17 mM Tris, pH 8.0, 50 mM NaCl, 0.3% Triton X-100, and a protease inhibitor cocktail tablet (Boehringer Mannheim). Specific proteins were detected using the following primary antibodies: γH2AX (Upstate), phospho-Ser1981 ATM (Rockland), and Chk2 (Cell Signaling). Immunoreactive bands were normalized to that of the control β-actin (Santa Cruz Biotechnology) band.
RESULTS
1) HDAC inhibition regulates the formation of H2AX foci
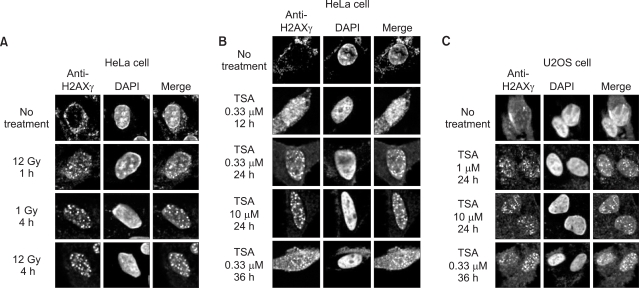
In this study, we examined whether TSA induces DNA damage, and whether the resultant signals differ from γ-irradiation-induced DNA damage signals. Phosphorylation of H2AX (γH2AX) has become an established marker of DNA damage, particularly double-strand breaks (DSBs). In response to DSBs, H2AX rapidly localizes to sites of DNA damage and is phophorylated in an ATM-dependent manner (21). Thus, to detect the dose-responsiveness of DNA damage, we checked the phosphorylation and foci of H2AX in response to γ-irradiation and TSA, respectively. As shown in Fig. 1A, H2AX foci were detectable at 1 h after exposure to 12 Gy irradiation. Formation of H2AX foci was maximal at 12 Gy at 4 h, and no additional foci were seen at 8 h, 12 h, 24 h, and 36 h after exposure to 1 Gy or 12 Gy irradiation (data not shown). These data suggested that the formation of H2AX foci is an early step in the cellular response to γ-irradiation.
We performed confocal analysis in order to estimate the time point of the formation of H2AX foci in response to TSA. The formation of H2AX foci was initiated in 0.33 µM TSA exposed HeLa cells for 12 h (Fig. 1B). The foci were maximal in 0.33 µM TSA at a time point of 24 h. Additionally, the formation of H2AX foci was initiated by exposure to 1 µM TSA for 24 h in U2OS cells (Fig. 1C). The H2AX foci were maximal upon exposure to 10 µM TSA, whereas the number of foci was decreased after exposure for 36 h. The dose of foci formation differs from cells to cells, but these foci were maximal at 24 h. Thus, these data suggested that the responses of TSA-induced DNA damage signals are more delayed than those induced by γ-irradiation.
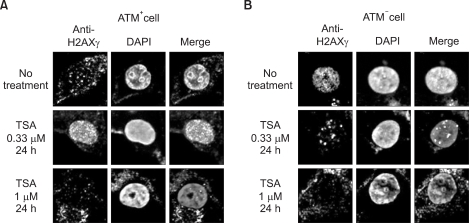
In addition, we examined the H2AX foci in ATM+ and ATM- cells treated with or without 0.33 µM and 1 µM of the HDAC inhibitor, TSA, for 24 h (Fig. 2). The H2AX foci were maximal upon exposure to 0.33µM TSA in ATM+ cells (Fig. 2A), whereas this was not shown in ATM- cells (Fig. 2B). From these data, we demonstrated that the H2AX foci can be formed by TSA in an ATM-dependent manner.
2) HDAC inhibition regulates the phosphorylation of H2AX
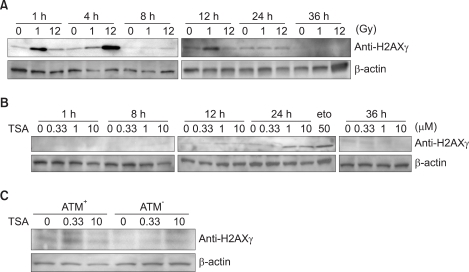
To examine the kinetics of H2AX phosphorylation, we performed immunoblotting using the γ-H2AX antibody (Fig. 3). As expected, phosphorylation of H2AX was mostly coincident with the results of the formation of foci in response to TSA (Fig. 1, 2). Phosphorylation of H2AX was observed as early as 1 h after exposure to 1 Gy irradiation (Fig. 3A). Phosphorylation of H2AX occurs within 12 h with kinetics that are similar to the kinetics of H2AX foci in response to DSBs.
H2AX is also phosphorylated in response to TSA (Fig. 3B). In contrast to γ-irradiation, the phosphorylation of H2AX was barely detected until 24 h. These results further support the conclusion that TSA induces DNA double-strand breaks after 24 h as a mechanism for the activation of ATM.
3) HDAC inhibition induces the phosphorylation of ATM
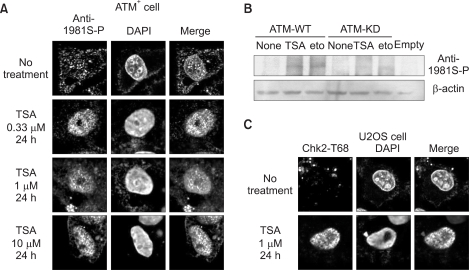
In response to DNA double-strand breaks and some other forms of stress, ATM becomes autophosphorylated at Ser1981 (17). To determine if stimulation of the ATM pathway by TSA might also involve autophosphorylation of ATM at Ser1981, phosphor-Ser1981 ATM antibody was used in confocal analysis and immunoblotting. In ATM+ cells, ATM was phosphorylated by 0.33 µM TSA for 24 h (Fig. 4). In contrast, ATM phosphorylation showed a much lower response in other doses compared with 0.33 µM TSA. This finding demonstrates that TSA phosphorylates ATM, and that it is an ATM-dependent DNA damage signal.
Similar results were obtained by immunoblotting (Fig. 4B). Phosphorylated ATM was also observed in 293T cells transfected with ATM-WT cells, but not ATM-KD cells treated with TSA for 24 h. As observed with ATM phosphorylation, the phosphorylation of Chk2 at Thr68 occurred in TSA-treated U2OS cells (Fig. 4C). These findings demonstrate that TSA could induce DNA damage signals that require ATM in each of the cell cultures used in these experiments.
DISCUSSION
Exposure of cells to DNA damaging stresses leads to the activation of complex signal transduction pathways that induce various critical cellular responses. Of these pathways, ATM-mediated signaling is an extremely important component of cellular responses to DNA damage, including cell cycle arrest, DNA repair, and apoptosis.
It is clear that ATM, a protein kinase, functions in diverse cellular processes in response to stresses by acting upon numerous protein substrates. Therefore, a functional deficiency of ATM affects many critical cellular processes, resulting in a pleiotropic disease phenotype in humans. The discovery that several ATM substrates participate in the DNA damage response pathways likely accounts for some of the abnormalities in DNA damage responses seen in A-T cells.
The chromosomal instability and cancer predisposition that characterize A-T are also observed in chromosome breakage syndromes such as Nijmegen breakage syndrome, Werner syndrome, and Fanconi anemia. Inhibition of HDAC induces chromosomal instability (7) and activates the cell cycle checkpoint (16) and ATM (17), which suggests that HDAC activity may be functionally implicated in DNA damage responses. In this study, we explored the effect of HDAC inhibition on activation of an ATM-mediated signaling pathway. We found that impaired histone acetylation/deacetylation activated an ATM-dependent DNA damage signal pathway. Thus, these results indicate that histone acetylation and deacetylation can act as different types of stresses that are transduced to the ATM signal pathway. In other words, altered chromatin remodeling can trigger an ATM-mediated DNA damage signal pathway, although further in vivo study is needed to assess the functional interplay between HDAC activity and the ATM-mediated DNA damage response.
Given that molecular responses to DNA damage can be modulated through altered HDAC inhibition following the activation of an ATM signal pathway by increased histone acetylation, we further question whether HDAC inhibitors can prevent carcinogenesis through the activation of DNA damage responses under the control of ATM. Indeed, cells can avoid or attenuate sickness if cells efficiently respond to stress-induced alterations when cells are exposed to stresses.
CONCLUSIONS
Based on our results showing that the alteration of chromatin remodeling can activate an ATM-mediated DNA signal pathway and can thereby result in diverse ATM-dependent responses, ATM might receive other signals in addition to its surrogate DNA damaging genotoxic signal. If this is the case, ATM can connect a variety of physiological pathways to each other, and induce functional interplay in response to many distinct types of stresses via the recognition and processing of distinct stress signals. Thus, ATM can play pleiotropic roles. In summary, as demonstrated here, ATM can respond to the altered histone acetylation/deacetylation (altered chromatin remodeling) through the activation of DNA damage signal transduction, via an ATM-Chk2 pathway. Thus, our study adds to our understanding of the signals for ATM activation.
 XML Download
XML Download