

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Cell cycle progression is regulated by checkpoint controls that function to protect the integrity of the genome. Such controls act to prevent cell cycle progression until after completion of prior events (1). The G1 checkpoint permits repair prior to replication, whereas arrest at the G2 checkpoint permits repair of the genome prior to its mitotic segregation. The p53 tumor suppressor gene, which is suppressed by mutation in approximately one-half of human tumors (2), has been shown to be an integral part of both the G1 (3) and G2 checkpoints (4).
Arrest at the G1 checkpoint results, at least in part, from p53-mediated synthesis of the cell cycle inhibitor p21, which binds to and inhibits the cdk-cyclin complexes such as cdk2-cyclin E and cdk2-cyclin A (5,6). However, there is controversy concerning the role of p53 in the G2 checkpoint. Since p53 knockout or mutant p53-expressing cells seem to arrest at G2/M following DNA damage (3), it was initially thought that p53 plays no role at the G2-M transition. However, our research group and others have reported that the expression of wild-type p53 in human cells with using a tetracycline-repressible promoter or the expression of the temperature-sensitive mutant form of p53 (p53val135) at a permissive temperature leads to an increase in the G2/M and G1 populations of the growth-arrested cells (7,8). We have also reported that constitutive activation of cyclin B-associated cdc2 kinase overrides p53-induced G2 cell cycle arrest (9).
It has been reported that p53 represses the transcription of such cell cycle-regulatory genes as cdc2 and cyclin B via the inactivation of NF-Y (10), which seems to cause an inactivation of cdc2 kinase and then cell cycle G2 arrest (9). Further, NF-Y has been suggested to be involved in transcriptional repression of G2-specific genes after p53 induction and also DNA damage (10,11). The DNA binding activity of NF-Y is decreased by p53 (12). Moreover, we have recently reported that cdk2 phosphorylates two serine residues near the DNA binding domain of the YA subunit of NF-Y (13). This phosphorylation appeared important for the DNA binding activity of NF-Y and the transcription of cyclin A, cyclin B, cdc2 and cdc25C (14).
Although p53 can inhibit cdc2 kinase through transcriptional repression of cdc2 and cyclin B, and cdc25C protein phosphatase (14), cdc2 kinase could be inhibited in a p53-independent manner by DNA damage-induced, ATM dependent phosphorylation of checkpoint kinases (Chk1 and Chk2), and they in turn phosphorylate and inactivate cdc25C. In this study, we investigated a role of p53 for the cell cycle G2 checkpoint. We found that p53 is critical for sustaining G2 checkpoint arrest, but not for arresting the cell cycle at the G2 checkpoint. Therefore, these findings suggest that a p53-dependent backup mechanism is critical to sustain G2 arrest until completion of repairing the DNA damage.
MATERIALS AND METHODS
1) Cell culture and transfection
Human HCT116 and its derivatives were obtained from Dr. Vogelstein (Johns Hopkins Medical Oncology Center, Baltimore, MD) and they were cultured in Dulbecco's modified Eagle's medium (DMEM) that was supplemented with 10% fetal bovine serum. The DNA transfections were performed using the CaPO4 coprecipitation procedure (15).
2) Cell cycle analysis
To analyze the DNA content by flow cytometry, we trypsinized the cells, fixed them in 70% ethanol, resuspended them in Dulbecco's phosphate-buffered saline that contained RNase A (50µg/ml), 0.1% sodium citrate, propidium iodide (50µg/ml) and 0.1% Triton X-100. We then analyzed the samples by a FACScan (Becton Dickinson, Mountain view, CA) and with using Lysys software (14).
3) Western blot analysis
The cell lysates were prepared as previously reported (10). Briefly, the cells were lysed with RIPA buffer [50 mM Tris (pH 7.5), 5 mM NaCl, 1µM EGTA, 1% Triton X-100, 50µM NaF, 10µM Na3VO4, 1µg/ml aprotinin, 1µg/ml leupeptin, 1µg/ml pepstatin A, 0.1 mM PMSF and 1 mM DTT], and the lysates were clarified by centrifugation (1,200 rpm for 10 min at 4℃). The total cell extracts were separated on SDS-polyacrylamide gel, and the proteins were transferred to a PVDF membrane (NEN Life Science Products, Boston, MA) Rabbit antibody against cyclin B1 (SC-752; Santa Cruz Biotechnology, Santa Cruz, CA), anti-cdc25C (SC-327; Santa Cruz Biotechnology, Santa Cruz, CA), and anti-RNR R2 (SC-10846; Santa Cruz Biotechnology, Santa Cruz, CA), and mouse antibodies against cdc2 (SC-54; Santa Cruz Biotechnology , Santa Cruz, CA), Flag (M2; Sigma-Aldrich, Saint Louis, MO) or p53 (SC-126; Santa Cruz Biotechnology, Santa Cruz, CA), and goat antibody against actin (SC-1616; Santa Cruz Biotechnology, Santa Cruz, CA) were used for the Western blot assay. Immunoblotting was performed with ECL Western blotting detection reagents (Amersham Pharmacia Biotech, Sweden).
4) In vitro kinase assay
The activities of cdc2 and cdk2 were measured by IP-Kinase. For the immunoprecipitation kinase assay (IP-kinase assay), the cells were washed with cold PBS and then lysed in RIPA buffer [50 mM Tris-HCl (pH 7.5), 5 mM NaCl, 1µM EGTA, 1% Triton X-100, 50µM NaF, 10µM Na3VO4, 1µg/ml aprotinin, 1µg/ml leupeptin, 1µg/ml pepstatin A, 0.1 mM PMSF, 1 mM DTT]. The extracts (200µg) were incubated for 12 h at 4℃ with 2µg of anti-cdk2 (SC-163; Santa Cruz Biotechnology, Santa Cruz, CA) and anti-cdc2 (SC-54; Santa Cruz Biotechnology, Santa Cruz, CA). The immunoprecipitates were immobilized on protein A-agarose beads (Boehringer Mannheim, Germany) by incubation for 4 h at 4℃. The beads were washed twice with 1 ml RIPA buffer and then twice with kinase buffer [50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM DTT]. Following the final wash, the immune complexes were suspended in 50µl of the corresponding kinase buffer that contained 20µM ATP, 5µCi [γ-32P]ATP and 2µg substrate, and specifically histone H1 (Boehringer Mannheim, Germany) for the cdk2 and cdc2 assays. The reactions were allowed to proceed for 30 min at 30℃. The phosphorylated proteins were separated on a 12% SDS-polyacrylamide gel and then visualized by autoradiography.
5) In vivo phosphorylation analysis
The cell were washed twice with TBS; this was followed by 4 hrs of labeling in 3 ml of phosphate-free Dulbecco's modified Eagle's medium and 10% dialyzed fetal bovine serum that contained 0.5 mCi of [32P]orthophosphate. The labeled cells were treated with 1 ml of Nonidet P-40 lysis buffer [50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM dithiothreitol, 0.5% NP-40, 50 mM NaF, 10µM Na3VO4, 1µg/ml aprotinin, 1µg/ml leupeptin, 1µg/ml pepstatin A and 0.1 mM PMSF]. The labeled lysates were immunoprecipitated with anti-FLAG antibody and protein A-agarose. After washes, the samples were boiled and loaded onto a 12% SDS-polyacrylamide gel for electrophoretic separation. The phosphorylated protein species were at last visualized for autoradiography.
RESULTS
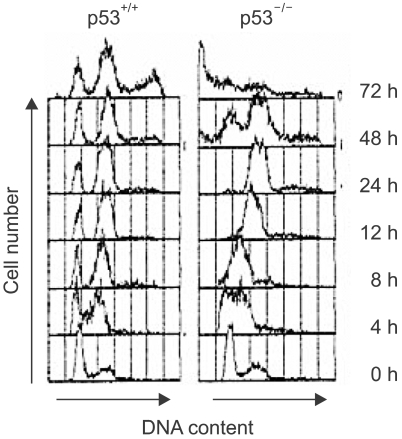
To compare the DNA damage-induced cell cycle arrests between the p53-positive cells and the p53-deficient cells, we examined the cell cycle of the human colon cancer cell line, that is, HCT116 cells and its p53-knockout derivative (p53-/-). After γ-ray irradiation, the parental p53-positive cells were arrested at the G1 and G2 phases, but the p53-deficient cells failed to arrest at G1, but they did become arrested at G2 (Fig. 1). However, while the cells with functional p53 sustained their cell cycle G1 and G2 arrests until 72 h, the p53-deficient cells entered mitosis at 48 h after the DNA damage and a sub-G1 population appeared at 72 h (Fig. 1). Therefore, these results imply that the p53-deificent cells inappropriately entered mitosis and this resulted in significant increases in the sub-G1 dead cell populations.
Since it has been reported that cdc2 kinase play a key role for entering mitosis, we examined the expression of cdc2 and cyclin B and also the activity of cdc2 kinase. The cyclin B expression was significantly decreased in the parental p53-positive cells, but not in the p53-dificient cells (Fig. 2A). There was a lesser decrease in the protein levels of cdc2 than that for cyclin B, but these levels were near the bottom levels at 48 h after irradiation. Of note, the highly phosphorylated forms of cdc2 disappeared in the p53-positive cells, but they were sustained until 24 h in the p53-deficient cells (Fig. 2A). Cdc2 kinase was inactivated after irradiation in both the p53-positive cells and the p53-deficient cells (Fig. 2B). However, the p53-deficient cells showed restored cdc2 kinase activity 24 h after irradiation and this cdc2 reactivation seemed to be associated with entry into mitosis by the p53-deficient cells. Therefore, these results imply that p53 is dispensable for G2 arrest in response to DNA damage, but p53 is indispensable for the maintenance of G2 checkpoint arrest via sustaining the inactivation of cdc2 kinase and repressing the cdc2 and cyclin B expressions.
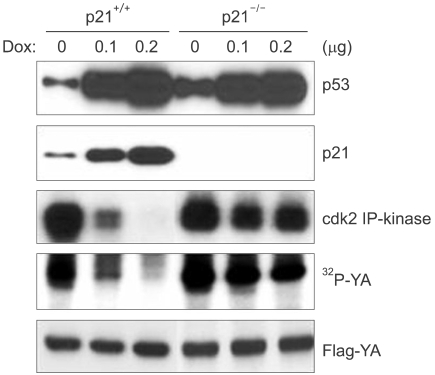
We next examined whether p21, which is a major mediator of p53 function in cell cycle regulation, affected the expressions of cyclin B and cdc2 following DNA damage. The p21-deficient cells (p21-/-), like the cells deficient for p53, also failed to repress the expressions of cdc2 and cyclin B following DNA damage (Fig. 3). In addition, other cell cycle regulatory genes such as cyclin A, RNR-R2 and cdc25C, which are all known to contain the NF-Y binding CCAAT motif in their promoter regions, were also decreased in the HCT116 cells in response to DNA damage, but this didn't occur in the p21-deficient cells (Fig. 3). Therefore, these results suggest that p21 mediates the functions of p53 that keep the cell in cell cycle G2 arrest following DNA damage. We previously reported that cdk2 phosphorylates the YA subunit of the NF-Y transcription factor and this phosphorylation is essential for DNA binding and the transactivation ability of NF-Y. Since the cell cycle regulatory genes, including cdc2, cdc25C, cyclin A, cyclin B and RNR-R2, are known to contain the NF-Y binding CCAAT motif in their promoter regions and their transcription is dependent to NF-Y, we examined for YA phosphorylation in the p53-deficient cells following DNA damage. The IP-kinase assay showed inactivation of cdk2 in the parental HCT116 cells upon DNA damage. However, in the p21-deficient cells, cdk2 was still active after DNA damage (Fig. 4). Furthermore, YA phosphorylation was decreased with cdk2 inactivation in the parental HCT116 cells, but this did not happen in the p21-deficient cells: this implies that p53 and its transactivation target p21 are necessary to dephosphorylate the YA subunit through the inhibition of cdk2.
DISCUSSION
In this study, we found that p53 is critical to sustain cell cycle arrest at the G2 checkpoint in DNA damaged cells, but it is not crucial to arrest cells at G2, which is indispensable for maintenance of cell cycle G2 arrest and for preventing inappropriate entry into mitosis. Since it is believed that cells re-enter the cell cycle from checkpoint arrest after completion of repairing the damaged DNA, p53-deficiency may cause genetic instability by allowing cells to enter mitosis from G2 arrest without completing their DNA repair.
In this study, we investigated a role for p53 in the cell cycle G2 checkpoint and we found that p53 is indispensable for sustaining G2 checkpoint arrest, but not for arresting the cell cycle at the G2 checkpoint. Therefore, these finding suggest that a p53-dependent backup mechanism is critical to sustain G2 arrest in DNA damaged cells. These findings also explain why the loss of p53 apparently sensitizes tumor cells to microtubule inhibitors and so leads to genomic instability.
CONCLUSIONS
p53 is necessary to sustain cell cycle G2 arrest by keeping cdc2 kinase inactive following DNA damage. We next examined whether p21, which is a major mediator of p53 functions in cell cycle regulation, affects the expression of cyclin B and cdc2 following DNA damage. The p21-deficient cells (p21-/-), like the cells deficient for p53, also failed to repress the expression of cdc2 and cyclin B following DNA damage. Also, p21 is necessary to decrease the cdk2-dependent phosphorylation of the YA subunit of the NF-Y transcription factor, which is essential for NF-Y-dependent transcription of such cell cycle-regulatory genes as cdc2 and cyclin B. p53 is essential to sustain cell cycle G2 arrest through the p21-cdk2-NF-Ysignaling pathway.


 XML Download
XML Download