

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Classically, angiogenesis is a process of new blood vessel formation from preexisting vessels (1). More specifically, angiogenesis is the mechanism mediating the growth and modification of a capillary network (2,3). Although normal endothelium is generally quiescent in the adult organism, angiogenesis is prominent during conditions of tissue repair and remodeling (eg, menstrual cycle and mammary gland involution), and pathologic events, such as wound healing, rheumatic diseases, diabetic retinopathy, inflammation, and malignancy (2,4).
Indeed, angiogenesis is an essential process in neoplastic growth. In fact, for a tumor to develop a highly malignant and deadly phenotype, it must first recruit and sustain its own blood supply (5). Since cells cannot survive if they lack an adequate supply of oxygen and nutrients, the expansion of a tumor mass beyond 1 mm in diameter is therefore dependent on the recruitment of its own vascular supply, by angiogenesis and/or blood vessel cooption (5~7). The process of angiogenesis is also sustained by secretion of specific angiogenic factors by cancer and non-cancer cells. Angiogenic factors can initiate a multi-step process that begins with vasodilatation, followed by the enhancement of vessel permeability and stroma degradation. Angiogenic factors can also promote endothelial cell migration and proliferation. As a result, endothelial cells converge and tightly assemble to generate a new lumen or they intercalate with endothelial cells in a preexisting vessel to give rise to capillary elongation. The newly formed capillary-like vessels undergo final remodeling, in which they rearrange from an irregular into a structured plexus of branching vessels and capillary loops (8). A hallmark of solid tumors is represented by abnormal vasculature. Tumor vessels are organized in a chaotic fashion, characterized by sinuous, extended, and enlarged vessels and are randomly fused with either arterioles or venules, creating an atypical microcirculation (9). In addition, tumor vessels are leaky, as a result of endothelial cell aberrant lining, fenestrations, vesicles and transcellular holes, widened inter-endothelial junctions, and a discontinuous or absent basement membrane (9~12). Tumor vessels may also lack functional perivascular cells (13) and their walls can be formed by a mosaic of alternating endothelial and cancer cells (vasculogenic mimicry), thereby exposing cancer cells to the lumen (14,15). As a result, neoplastic regions are often hypoxic and acidic due to the chaotic and slow blood flow (2,16~19). Tumor vessels infiltrate the neoplastic mass, sustaining its growth and its metastatic perfusion.
The abnormal feature of the tumor vasculature is likely to represent an imbalanced expression of angiogenic factors and inhibitors within a tumor. The blood vessel growth in normal tissues is regulated by the action of angiogenic stimulators, "proangiogenic factors" and angiogenic inhibitors, "antiangiogenic factors". The stimulators include: basic fibroblast growth factor (FGF-2), a potent mitogen for endothelium, VEGF, the most potent endothelial chemoattractant; cyclooxygenase 2 (COX-2), matrix metalloproteinases (MMPs), and transforming growth factor-beta 1 (TGF-beta1). Antiangiogenic factors counteract the effect of the angiogenic stimulators and therefore keep the normal blood vessels growth under a strict and well balanced control. These antiangiogenic factors include tissue inhibitor of metalloproteinase 1 (TIMP-1), interleukin-10 (IL-10), angiostatin, endostatin, and interferon (IFN) (20). Thus, activation of the angiogenic switch during early stages of tumor development suggests that regulation of the angiogenic process is likely to be a rate-limiting step in the progression from small lesion to extensive disease (3,5,21).
FIBROBLAST GROWTH FACTORS AND FIBROBLAST GROWTH FACTOR-BINDING PROTEIN
The FGF family comprises to date more than 20 distinct, structurally-related proteins that exert biologic effects in different cells and organ systems, including tumor growth and angiogenesis (22~25). FGFs are heparin-binding proteins, which interact with low affinity heparan sulfate proteoglycans (HS-PGs). HSPGs can protect FGFs from thermal denaturation and proteolysis and can increase FGF receptor affinity and facilitate FGF binding to cell surface receptor. In addition, ECM-associated HSPGs modulate FGF bioavailability by generating a local reservoir for the growth factor and allowing a sustained stimulation of endothelial cells (22,24). One mechanism that explains how these FGFs are mobilized from the extracellular storage includes the action of heparinases or other glycosaminoglycan-degrading enzymes (22,24). An independent mechanism is the action of a secreted FGF binding protein (FGF-BP) that functions as an extracellular chaperone for the locally-stored FGFs. Interestingly, FGF-BP is likely to complement the role of heparan sulfate in mediating FGF-dependent signaling and mitogenesis (26~28). FGF-BP was first described in 1991 by Wu et al as a low-affinity heparin binding protein, isolated from A431 human epidermoid carcinoma cells (29). FGF-BP binds to FGF-1 and FGF-2 in a noncovalent, reversible manner facilitating the release of the growth factor from the ECM and presenting it to FGF receptors (27,30~32). Numerous studies conducted in our laboratory have confirmed a role for FGF-BP as an extracellular chaperone for FGFs that enhances FGF-dependent biochemical and biologic functions. In murine NIH-3T3 fibroblasts as well as in bovine GM7373 endothelial cells, FGF-BP was shown to enhance FGF-2-induced MAPK signaling pathway and cell proliferation and we showed recently that the interaction domain with FGF-2 is a short domain in the C-terminus of the FGF-BP protein (26,27,33). Furthermore, FGF-BP was responsible for enhancing FGF-2-mediated angiogenesis in a chick chorioallantoic membrane (CAM) assay (27). Overexpression of FGF-BP in a chicken transgenic model resulted in embryonic lethality due to massive disruption of blood vessel structure and integrity and subsequent hemorrhage (34).
FGF-BP is expressed below the level of detection by Northern blotting in normal adult human tissues. However, its expression is considerably elevated in various tumors, including skin, head and neck, cervical, lung, squamous cell carcinomas, breast, colon, and pancreatic adenocarcinomas (30,31,35~37). Expression in FGF-BP-negative cell lines indicated FGF-BP as a rate- limiting factor for tumor growth and angiogenesis in a nude mouse model (30). Similarly, ribozyme-targeting and depletion of endogenous FGF-BP from human squamous cell carcinoma and colorectal cancer cell lines resulted in a significant reduction of tumor growth and angiogenesis. These findings support a potential role of FGF-BP as an angiogenic switch in human neoplasia (31).
ANGIOGENESIS IN COLORECTAL AND PANCREATIC ADENOCARCINOMAS
1) Colorectal cancer
Colorectal cancer is the third most common type of cancer among men and women in the United States. Over 95% of colorectal cancers are adenocarcinomas. The other 5% is represented by other, more rare, types of colorectal neoplasias. Eighty-five percent of colorectal cancer patients undergo surgical removal of the primary tumor. Although early stages of the disease (Duke's A and B) are linked with a good post-operative prognosis and 80~95% of remission rate, tumor cell infiltration through the serosa (B2 stage) and lymph node metastasis (C stage) reduce the 5-year survival to 25% to 60% (38). The American Cancer Society estimates that there will be approximately 148,000 new cases of colorectal cancer and 55,000 deaths in the 2006 in the United States. Risk factors include age, a diet rich in fat and cholesterol, ethnic background, alcohol assumption, inflammatory bowel disease (ulcerative colitis, Crohn's disease), and family history, including hereditary polyposis and nonpolyposis syndromes. Reductions in morbidity and mortality can be achieved through detection and treatment of early-stage colorectal cancers and the identification and removal of adenomatous polyps. Colorectal cancer screening tests, such as fecal occult blood test (FOBT), fecal himmunochemical test (FIT), flexible sigmoidoscopy, and colonoscopy, performed in asymptomatic 50 or more year old individuals, allows accurate detection of early stage cancerous and precancerous lesions.
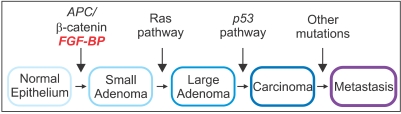
Pioneering research by Vogelstein and colleagues conducted in the past 15 years has significantly contributed in the understanding of the molecular basis of colorectal cancer initiation and progression (39). Unlike other types of malignancies, colon adenocarcinomas develop through a progression of clinical and histopathologic processes that proceed from a single crypt lesion through small benign tumors (adenomatous polyps) to malignant and invasive carcinomas (40) (Fig. 1). The earliest genetic alteration promoting the initiation of colorectal malignant transformation is a germline mutation in the adenomatous polyposis coli (APC) tumor suppressor gene (39,41,42) that generates a truncated APC protein unable to interact and suppress beta-catenin activity (43~49). The resulting sustained beta-catenin overexpression promotes the activation of growth-promoting oncogenes, such as cyclin D1 or c-myc (50~52). Interestingly, mice carrying an endogenous APC mutation (ApcMin/+) similar to that found in human colorectal cancers do not survive beyond 4 months of age as a result of development of adenomatous polyps that rapidly progress to colorectal malignancies (53). Mutation of the ras proto-oncogene has been detected in various human cancers, including 95% of pancreatic cancers and up to 50% of large bowel adenocarcinomas (54~56). Since this mutation has been identified in both small and large colon adenomas and also in adenocarcinomas (57~59), it is likely to represent a crucial step contributing to the transition from intermediate to late adenoma or adenocarcinoma (60). Chromosome arm 18q deletions, resulting in the mutation and reduced expression of DCC (deleted in colorectal carcinoma) tumor suppressor gene (61~64) and SMAD4/DPC4 (deleted in pancreatic carcinoma 4) (65~67) accounts for a later event associated with colon cancer progression through the stages of late adenoma to carcinoma (68). Loss or inactivation of p53 tumor suppressor gene, reported in a high percentage of colorectal cancers, is likely to be the latest event during disease progression (69). Disruption of p53 by gene targeting in human colon cancer cells results in cell resistance to different chemotherapeutic agents (70). Therefore, loss of p53 in human colorectal cancers may account for the inefficacy of chemotherapy and decreased patient survival (71~74).
In colorectal cancer, several studies indicate angiogenesis as a crucial event leading to colon cancer progression. As a matter of fact, colorectal cancer is one of the best-studied models of tumor angiogenesis (91). As in many other tumors, several angiogenic regulators have been recognized in colon cancer, including VEGF, PDGF, thrombospondin, and angiopoietins (91,92). Likewise, overexpression of FGF and FGFRs in colon cancer cells and tissues, as well as increase of FGF-2 serum levels in patients with advanced colon cancer, have been extensively reported (93~98).
2) Pancreatic cancer
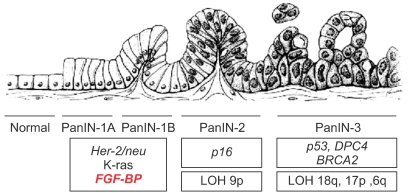
Pancreatic cancer is a cause of death of about 30,000 individuals each year in the Unites States (75). Although pancreatic cancer is recorded in only 2% of new cancer patients, it is the fifth leading cause of cancer-related death. Due to the lack of an efficacious early diagnostic test and to the manifestation of symptoms during late-stage disease, the malignancy is generally diagnosed after invasion and metastasis in surrounding tissues disabling patients to undergo curative resections. Another element playing a role in poor prognosis is the pancreatic cancer cell resistance to cytotoxic agents and radiation (76,77). Pancreatic adenocarcinoma develops through a step-wise progression from distinct epithelial lesions in the small interlobular ducts, namely pancreatic intraepithelial neoplasias (PanINs). PanINs can be flat (PanIN-1A), papillary without atypia (PanIN-1B), papillary with atypia (PanIN-2), or with characteristics of carcinoma in situ (PanIN-3) (78) (see Fig. 2). The molecular genetics of pancreatic adenocarcinoma have been well studied. Of these tumors, 80~95% have mutations in the K-ras gene (79,80), and 85~98% have mutations, deletions, or hypermethylation in the p16 tumor suppressor gene. Of these cancers, 50% have mutations in p53 and about 55% have homozygous deletions or mutations of DPC4/Smad4 and BRCA2. Some of these mutations can also be found in high-risk precursors of pancreatic cancer. For example, in chronic pancreatitis, 30% of patients have detectable mutations in p16 and 10% have K-ras mutations (81).
Although pancreatic cancer is not a grossly vascular tumor, it is often characterized by enhancement of tumor-dependent angiogenesis (82). A growing line of evidence has shown that various FGFs, such as FGF-1, FGF-2, FGF-5, FGF-7 (83~86) and FGF receptors (87~90) are upregulated in pancreatic cancer tissue samples and cell lines. These findings suggest that FGF-dependent downstream biologic events are likely to play an important role in the pathobiology of pancreatic cancer.
FIBROBLAST GROWTH FACTOR-BINDING PROTEIN IS AN EARLY MARKER IN GASTROINTESTINAL CANCERS
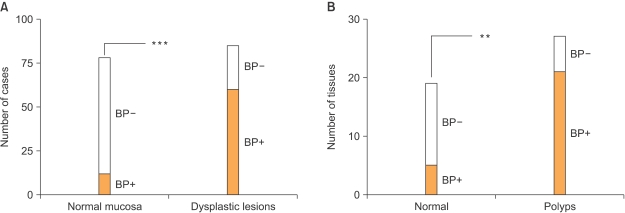
Our laboratory has recently focused on the analysis of FGF-BP expression and regulation during the progression of gastrointestinal cancers. In an initial study by Ray and colleagues (36), immunohistochemical analysis of normal and pathologic human colon biopsies using a polyclonal anti-FGF-BP antibody showed a significant upregulation of FGF-BP in dysplastic lesions, originating at the level of single crypts. Upregulation of FGF-BP protein was detected in a small portion (16%) of 76 apparently normal colon samples. However, expression of FGF-BP was detected in most samples with moderate to severe dysplasia (62 of 85; p<0.0001 normal versus dysplastic mucosa) (Fig. 3A). Furthermore, coincident with FGF-BP overexpression in dysplastic crypts, an increase of blood vessel density was found by immunostaining of the lamina propria with a CD31 antibody (from 80±7 to 154±9 vessels/field). The fact that specimens from different inflammatory bowel diseases, such as ulcerative colitis or Crohn's disease, did not show a significant FGF-BP upregulation prompted the authors to link FGF-BP overexpression in the onset of colon cancer to an early genetic occurrence. Analysis of FGF-BP mRNA by ISH in APCMin/+ mouse colon specimens showed a significant upregulation of FGF-BP in colon adenomas (21 of 27, >30% of the adenoma surface area). In contrast, little if any FGF-BP expression was noted in adjacent normal intestine (5 of 19; p<0.001, normal versus adenomas) (Fig. 3A). Interestingly, a prominent correlation between FGF-BP upregulation and cytoplasmic and nuclear beta-catenin staining in dysplastic colon was seen but not in a dextran sulfate-induced model of inflammatory colon disease (36). This finding further corroborated the hypothesis that FGF-BP is likely to be an early response gene during the initiation of colon cancer. Moreover, based on these findings and promoter-reporter studies, FGF-BP was shown to be a direct target of the activation of the Wnt/beta-catenin pathway (Fig. 1) and therefore represents an early event for colon cancer initiation.
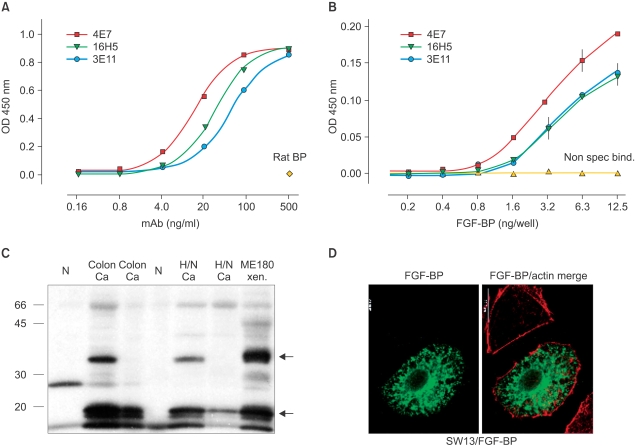
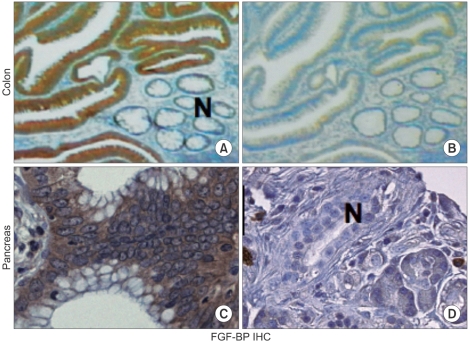
Due to the potential significance of this protein in different human cancers, we generated monoclonal antibodies (mAbs) to FGF-BP and employ them to identify of FGF-BP protein in various bioassays, such as ELISA, western blot, immunofluorescence (Fig. 4) and himmunohistochemistry (Fig. 5) (37). Monoclonal antibodies were applied for immunohistochemical (IHC) detection of FGF-BP in tissue microarrays containing a series of tissue samples from different stages of both colon and pancreatic cancers. In parallel, detection of FGF-BP mRNA in the same tissue slides was performed by in situ hybridization. High expression levels of FGF-BP protein and mRNA were found in adenomatous polyps (Fig. 5) and the upregulation was sustained throughout all other stages of colorectal cancer progression, such as invasive adenocarcinoma and metastases, relative to the levels detected in normal specimens (p<0.0001). In addition, FGF-BP protein and mRNA were found dramatically upregulated during the early stages of pancreatic cancer progression, such as low-grade (PanIN1 and 2) and high-grade PanIN as well as invasive adenocarcinoma. Positive FGF-BP expression was also evidenced in some pancreatitis specimens, whereas very low or no expression was detected in normal pancreatic ducts. Nonadenocarcinoma samples showed expression of FGF-BP in 23% of samples at low levels, although this expression still represented a significant increase relative to normal specimens (p=0.0044). A highly significant correlation resulted from comparison between protein and mRNA expression in all samples analyzed (37).
Based on these observations, it is tempting to speculate that FGF-BP is an early regulatory gene, activated during the early phases of colon and pancreatic cancer progression. It is also possible that, as a consequence of FGF-BP overexpression in the early stages of colon and pancreatic cancers, an enhancement of tumor-dependent angiogenesis and invasion may occur, due to the increase of FGF-BP-mediated FGF bioavailability.
Although the primary purpose of generating mAbs to FGF-BP was to generate better detection tools, we sought to also assess the potential of any of these mAbs to inhibit the function of FGF-BP in a bioassay. For this we used a previously established in vitro model (30) and identified the one monoclonal Antibody (mAb) as a blocking antibody that reduces anchorage- independent growth dependent on FGF-BP (37) (Fig. 6). This suggests these antibodies could be used to therapeutically target FGF-BP in cancer patients.
ANTIANGIOGENIC THERAPIES
Anti-angiogenic inhibitors are used as part of therapeutic strategies addressed to arrest endothelial cell proliferation and differentiation in the tumor environment, thereby preventing cancer growth and metastasis. In tumor masses, one capillary vessel with a single endothelial cell is able to provide nutrients to approximately 100 cancer cells. For this reason, inhibition of endothelial cell growth might represent an amplification factor in halting the progression of tumor growth. In addition, since physiological angiogenesis is downregulated in adults, minimal side effects are obtained upon prolonged anti-angiogenic treatments (100,101). Neutralization of pro-angiogenic factors expression or activity, inhibition of the capillary basement membrane turnover, and inhibition of endothelial cell proliferation, migration, and differentiation are the diverse effects that the therapeutic application of specific anti-angiogenic agents can target.
New antiangiogenesis therapies are emerging. In February 2004, the United States Food and Drug Administration (US-FDA) approved Bevacizumab (Avastin), the first angiogenesis drug for first line treatment of patients with metastatic colorectal cancer. Avastin is a humanized variant of monoclonal antibody (93% human, 7% mouse) that binds to VEGF with high affinity and neutralizes all VEGF-A isoforms, thereby blocking VEGF receptor binding. Due to the abnormal features of tumor vessels, angiogenic inhibitors should normalized these tumor vessels, in order to achieve better cytostatic drug delivery within the tumor tissue (102). For instance, prevention of vascular leakage would decrease the interstitial pressure within the tumor mass and increase blood perfusion and chemotherapeutic drug delivery when antiangiogenesis drugs and chemotherapeutic agents are combined. Indeed, the administration of Avastin with chemotherapeutic drugs has resulted in a significant improvement of median survival, free survival, percentage of response rate and median duration of response (103,104). Analogous clinical benefits were obtained in patients affected by non-squamous non-small cell lung cancer (NSCLC), breast and renal cancer when treated with a combination of Avastin and chemotherapy (105). It is then possible that the antitumor activity exerted by VEGF antagonists in combination with cytostatic agents results in an ameliorated VEGF-induced leakage and interstitial pressure and therefore in a more efficient cytostatic drug penetration (106).
CONCLUSION
Early events that initiate the stepwise progression of premalignant lesions of colon epithelia and pancreatic ducts to invasive cancer may provide circulating markers for the detection of such lesions. FGF-BP, as a secreted protein, could be such a marker to identify subjects at an increased risk for developing invasive cancers. Therefore, generation of a sensitive ELISA assay able to detect FGF-BP in patient sera may represent an important diagnostic screening method for early detection of colon and pancreatic premalignant lesions, thereby allowing early medical intervention.

 XML Download
XML Download