

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
A substantial body of evidence suggests that chronic inflammation acts as a driving force for the development of cancer (1). However, the cellular and molecular mechanisms that underlie the tumor-promoting effect of inflammation have not been fully elucidated. One of the frequently used animal models to study the mechanistic link between inflammation and the development of cancer is the classical two-stage mouse skin tumorigenesis model that involves an initiation and promotion scheme. According to this model, initiation of tumorigenesis is achieved by topical application of a relatively low dose of a genotoxic carcinogen such as 7,12-dimethylbenz[α]anthracene (DMBA), whereas promotion can be induced by repeated applications of a tumor promoter, including 12-O-tetradecanoylphrobol 13-acetate (TPA). Several previous studies have supported the concept of a tumor-promoting role for inflammation in mouse skin carcinogenesis (2~4). It has been reported that the transgenic overexpression of a pro-inflammatory enzyme cyclooxygenase-2 (COX-2), which is involved in the biosynthesis of prostaglandins (PGs) from arachidonic acid, sensitizes mouse skin for promotion of tumor (3). On the other hand, genetic ablation of cox-2 protects against chemically induced mouse skin tumorigenesis (4). Several studies have suggested that inflammatory mediators including the different PGs (e.g., PGE2, PGF2α and 15-deoxy-Δ12,14-PGJ2) are functionally related to mouse skin tumor promotion (2,5).
Topical application of TPA has been shown to induce the expression of COX-2 and its mRNA transcript in mouse skin (6) by activating the intracellular signaling that's mediated by various transcription factors and a series of upstream kinases. The 5'-flanking region of the cox-2 gene contains a canonical TATA box and the binding sequences for various transcription factors, including nuclear factor-kappaB (NF-κB), CCAAT/enhancer binding protein (C/EBP), cyclic AMP response element binding protein-binding protein (CREB), activator protein-1 (AP-1), etc. Inappropriate activation of these transcription factors often leads to the overexpression of cox-2.
Accumulating evidence from clinical, epidemiological and laboratory-based studies has suggested that non-steroidal anti-inflammatory drugs (NSAIDs) are effective in preventing certain cancers. Celecoxib, a selective COX-2 inhibitor developed for the treatment of inflammatory ailments, has been reported to reduce the burden of polyps in patients with familial adenomatous polyposis (7). Moreover, celecoxib inhibits experimentally induced tumorigenesis in several animal models (reviewed in 8, and see references therein), including azoxymethane-induced colon tumorigenesis in F344 rats, DMBA-induced rat mammary carcinogenesis, ultraviolet (UV) radiation-induced mouse skin tumor formation and N-butyl-N-(4-hydroxybutyl) nitrosamine-induced urinary bladder carcinogenesis in male B6D2F1 mice and female Fischer 344 rats. We have recently reported that topical application of celecoxib downregulates the TPA-induced COX-2 expression in mouse skin by blocking the activation of AP-1 via inactivation of the upstream p38 mitogen-activated protein (MAP) kinase (9). The present study was aimed to investigate the effects of celecoxib on TPA-induced tumor promotion and to determine the underlying molecular mechanisms in mouse skin in vivo.
MATERIALS AND METHODS
1) Materials
Celecoxib (Celebrex™) was supplied from Pharmacia Korea. TPA was purchased from Alexis Biochemicals (San Diego, CA). Rabbit polyclonal COX-2 antibody was procured from Cayman Chemical Co. (Ann Arbor, MI). Primary antibodies for vascular endothelial growth factor (VEGF), C/EBPδ, C/EBPβ and p65/RelA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin antibody was purchased from Sigma Chemical Company (St Louis, MO). Anti-rabbit and anti-goat horseradish peroxidase conjugated-secondary antibodies were the products of Zymed Laboratories (San Francisco, CA). The enhanced chemiluminescence (ECL) detection kit and [γ-32P]ATP were purchased from GE Healthcare (Buckinghamshire, UK).
2) Animal treatment
Female ICR mice (6~7 weeks of age) were purchased from the Dae-Han/Biolink Experimental Animal Center (Daejeon, Korea). The animals were housed in climate-controlled quarters (24℃ at 50% humidity) with a 12 h light/12 h dark cycle. The dorsal side of their skin was shaved using an electric clipper, and only those animals in the resting phase of the hair cycle were used in all experiments. TPA (10 nmol) and the respective doses of celecoxib were dissolved in 0.2 ml acetone and topically applied to the dorsal shaven area.
3) Two-stage mouse skin carcinogenesis
Female ICR mice were randomly divided into four groups, each consisting of 25 animals. Mice belonging to groups II, III and IV were treated with DMBA (0.2µmol) dissolved in 0.2 ml acetone : DMSO (85 : 15, v/v) on their shaven backs. The control mice in group I were treated with the same volume of solvent. One week after the initial treatment with DMBA, TPA (10 nmol/0.2 ml acetone) was topically applied to the animals in groups II, III and IV twice a week until termination of the experiment. Celecoxib was topically applied 30 min before each of the TPA treatments in groups III and IV at a dose of 1 and 10µmol, respectively. Starting at 1 week following treatment with the promoter, tumors of at least 1 mm diameter were counted every week until the termination of the experiment. The results were expressed as the percentage of papilloma-bearing mice (incidence) and the average number of papillomas per mouse (multiplicity).
4) Western blot analysis
The mouse skin papillomas were collected, incised and washed with ice-cold phosphate-buffered saline (PBS). In other short-term experiments, the shaven backs of the female ICR mice were topically treated with celecoxib (1 or 10µmol) 30 min before application of TPA (10 nmol); then they were killed by cervical dislocation either 1 or 4 h later. The collected papillomas or epidermis were homogenized in 800µl of ice-cold lysis buffer [150 mM NaCl, 0.5% Triton-X 100, 50 mM Tris-HCl (pH 7.4), 20 mM EGTA, 1 mM DTT, 1 mM Na3VO4 and a protease inhibitor cocktail tablet]. The lysates were centrifuged at 14,800×g for 15 min. The supernatant was collected and the total protein concentration was quantified by using a BCA protein assay kit. The cell lysates (30~50µg protein) were boiled in SDS sample buffer for 5 min before electrophoresis on 10~12% SDS-polyacrylamide gel. After transfer to a PVDF membrane, the blots were blocked with 5% fat-free dry milk-PBST buffer (PBS containing 0.1% Tween 20) for 1 h at room temperature and then they were washed with PBST. The membranes were incubated for overnight at 4℃ with 1 : 1,000 dilutions of the primary antibodies for COX-2, VEGF, C/EBPδ and C/EBPβ, and also with a 1 : 500 dilution of p65/RelA. The blots were washed three times with PBST at 5-min intervals followed by incubation with an 1 : 5,000 dilution of the respective horseradish peroxidase conjugated-secondary antibodies (rabbit or goat) for 1 h, and then they were again washed in PBST for three times. The transferred proteins were visualized with an ECL detection kit according to the manufacturer's instructions.
5) Preparation of nuclear extracts from the mouse skin
The nuclear extract from mouse skin was prepared as previously described (6). In brief, the scraped dorsal skin was homogenized in 800µl of hypotonic buffer A [10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 1 mM DTT, 0.1 mM EDTA and 0.1 mM phenylmethylsulfonylfluoride (PMSF)]. 80µl of 10% Nonidet P-40 (NP-40) solution was added to the homogenates, and the mixtures were then centrifuged for 2 min at 14,000×g. The supernatant was collected as a cytosolic fraction. The precipitated nuclei were washed once with 500µl of buffer A plus 40µl of 10% NP-40, centrifuged, resuspended in 200µl of buffer C [50 mM HEPES (pH 7.8), 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1 mM PMSF and 20% glycerol); then they were centrifuged for 5 min at 14,800×g. The supernatant containing nuclear proteins was collected and stored at -70℃ after determination of the protein concentrations.
6) Electrophoretic mobility shift assay (EMSA)
The EMSA for DNA binding of the different transcription factors was performed by using a DNA-protein binding detection kit according to the manufacturer's protocol (GIBCO BRL, Grand Island, NY). Briefly, oligonucleotides harboring the binding sites for NF-κB (5'-GATCGAGGGGGACTTTCCCAGC-3') and C/EBP (5'-AGAGATTGCCTGA CGTCAGAGAGCTAG-3') were labeled with [γ-32P] ATP by using T4 polynucleotide kinase, and these oligonucleotides were purified on a Nick column (GE Healthcare, UK). The binding reaction was carried out in 25µl of a mixture containing 5µl of incubation buffer [10 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 4% glycerol and 0.1 mg/ml sonicated salmon sperm DNA], 10µg of nuclear extracts and 100,000 cpm of the [γ-32P]ATP-labeled oligonucleotide. After 50 min incubation at room temperature, 2µl of 0.1% bromophenol blue was added, and the samples were electrophoresed through 6% non-denaturing polyacrylamide gel at 150 V in a cold room for 2 h. The gel was finally dried and exposed to X-ray film.
RESULTS
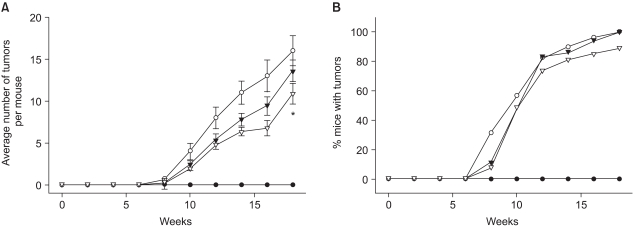
1) Suppression of DMBA-initiated and TPA-promoted mouse skin tumorigenesis by celecoxib
Our previous study demonstrated the suppression of the TPA-induced COX-2 expression by celecoxib (9), which prompted us to examine the effect of this COX-2 specific inhibitor on the promotion of mouse skin tumor. Celecoxib (1 or 10µmol) was applied to the DMBA-initiated dorsal skin of female ICR mice 30 minutes prior to each topical application of TPA for twice a week for 18 weeks. The onset of papillomas in the DMBA-initiated mouse skin occurred 8 weeks after TPA treatment (Fig. 1). At the 18th week, all the animals in the DMBA plus TPA-treated group developed papillomas at about 18 tumors/mouse. Pretreatment with celecoxib at a dose of 10µmol significantly (p<0.05) lowered the multiplicity of the papillomas (Fig. 1A), while the tumor incidence was only slightly reduced (Fig. 1B).
2) Inhibitory effects of celecoxib on the expression of COX-2 and VEGF in mouse skin papillomas
Since COX-2 has been implicated in tumorigenesis (8), we examined the effect of celecoxib on the expression of COX-2 in mouse skin papillomas. Our study revealed that the expression level of COX-2 was elevated in the papillomas collected from the DMBA plus TPA treated group. Celecoxib (10µmol) pretreatment significantly (p<0.01) reduced the expression of COX-2 in the papillomas, while the expression of the housekeeping enzyme COX-1 remained largely unchanged (Fig. 2A, 2C).
An elevated expression of COX-2 has also been implicated in angiogenesis, which is a cardinal feature of the rapid growth of a tumor mass (10,11). One of the principal angiogenic factors is VEGF, which is a potential target of various anticancer agents (12). In the present study, we sought to examine the effect of celecoxib on the expression level of VEGF in mouse skin papillomas. As shown in Fig. 2B and 2C, mouse skin papillomas from the celecoxib pretreated group exhibited a significant (p<0.01) decrease in the expression of VEGF as compared to that observed in the papillomas from animals treated with only DMBA plus TPA.
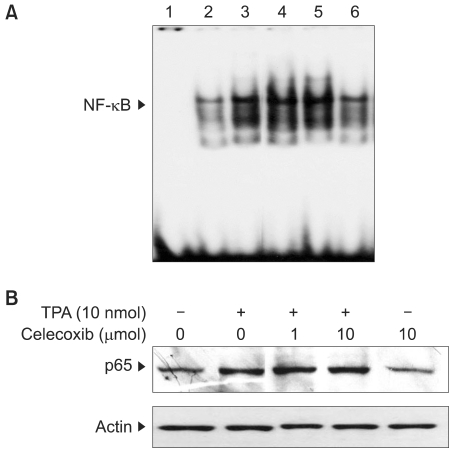
3) Effects of celecoxib on the activation of NF-κB in TPA-stimulated mouse skin
We have recently reported that celecoxib inhibits COX-2 expression by blocking the activation of an eukaryotic transcription factor AP-1 via the down-regulation of upstream p38 MAP kinase in TPA-stimulated mouse skin (9). Besides AP-1, NF-κB has been shown to play a key role in regulating the expression of COX-2. Therefore, we attempted to examine the effect of celecoxib on TPA-induced activation NF-κB in mouse skin in vivo. Our study revealed that a single topical application of celecoxib (10µmol) failed to affect TPA-induced nuclear translocation of an NF-κB subunit protein p65/RelA (Fig. 3A) and the DNA binding of NF-κB in mouse skin (Fig. 3B).
4) Inhibitory effects of celecoxib on TPA-induced C/EBP activation in mouse skin
To further identify the molecular targets of celecoxib for suppressing tumor promotion and the COX-2 expression in TPA-treated mouse skin, we examined the effect of celecoxib on the activation of another transcription factor C/EBP (NF-IL6), which is known to regulate the COX-2 expression (13). Pretreatment with celecoxib (10µmol) diminished the TPA-induced DNA binding of C/EBP (Fig. 4A). Celecoxib also attenuated the nuclear expression of C/EBPδ, but not C/EBPβ, in the TPA-stimulated mouse skin (Fig. 4B).
DISCUSSION
Chemoprevention is an innovative approach that focuses on pharmacological and nutritional interventions to prevent, reverse or delay carcinogenesis. The concept of chemoprevention by targeting inflammation has recently been promulgated through multiple lines of evidence from preclinical and clinical studies (1,14). A wide spectrum of anti-inflammatory substances, especially selective COX-2 inhibitors, have been shown to prevent the onset or progression of various cancers (7,8,14). Thus, prolonged intake of aspirin and other NSAIDs have been reported to reduce the risk of cancer of different organs including the colon and other gastrointestinal organs, breast, prostate, lung and skin. Although there is controversy that the cardiovascular toxicity of selective COX-2 inhibitors (15) may offset the benefits of these agents in cancer chemoprevention, recent clinical studies have demonstrated that not all COX-2 inhibitors are equivalent for provoke cardiac toxicity (16,17). Moreover, the European Medicines Agency has concluded that COX-2 inhibitors are contraindicated only for those patients with established cardiovascular disorders and so they should be used with caution in patients with risk factors (18). A nested case-control study on a cohort of 469,674 patients revealed that the use of rofecoxib and etoricoxib, but not celecoxib, was associated with a significantly increased risk of ischemic stroke (19). Moreover, a recent cohort study conducted among 74,838 subjects demonstrated that the risk of cardiovascular events was significantly elevated with the use of rofecoxib and it was significantly reduced with naproxen, while the use of celecoxib did not cause a significant increase or decrease in the cardiovascular event rate (15).
There is now ample evidence suggesting that one of the epigenetic mechanisms involved in the field of carcinogenesis is the overamplification of the inflammatory signaling that results in aberrant COX-2 induction (8,20). Therefore, COX-2 has been recognized as a molecular link between inflammation and cancer (8,20). To explore the molecular basis of the antitumor promoting effect of celecoxib, we have examined the effect of celecoxib on the expression of COX-2 in mouse skin papillomas. In the present study, the papillomas produced by DMBA plus TPA treatment exhibited a significant elevation of their COX-2 expression levels, which was diminished by celecoxib pretreatment during the promotion stage. Therefore, it is plausible to infer that COX-2 is a valid molecular target of celecoxib for achieving an antitumor-promoting effect with using this selective COX-2 inhibitor. Our previous studies demonstrated that single topical application of celecoxib down-regulated the COX-2 expression in mouse skin that had been treated with a single dose of TPA (9). Celecoxib, like the other NSAIDs, has been developed as a drug that can inhibit COX-2-catalyzed reactions. Besides down-regulating the COX-2 expression in mouse skin, celecoxib that is topically applied at the same dose (10µmol) may also inhibit the catalytic activity of this enzyme as well. However, multiple COX-2-independent mechanisms that underlie the anticarcinogenic effects of celecoxib have also been documented (21).
The growth of a solid tumor involves extensive neovascularization, which is promoted by an elevation of VEGF. Skin papillomas from the DMBA plus TPA treatment group showed an elevated expression of VEGF, but this was suppressed in the papillomas from the celecoxib pretreated group; this suggests that COX-2 plays an important role for angiogenesis during the formation of mouse skin papilloma. Similar to our findings, the antiangiogenic effect of celecoxib was evidenced by the decreased expression of VEGF in human pancreatic cancer cells (22) and the retinas of streptozotocin-induced diabetic rats, as well as the reduced secretion and mRNA levels of VEGF in human retinal pigment (ARPE-19) cells (23). Further investigation is necessary for verifying the reduced blood vessel formation in the papillomas from celecoxib-treated mouse skin.
We have previously reported that celecoxib inhibited the TPA-induced COX-2 expression in mouse skin by blocking the activation of AP-1 via the modulation of p38 MAP kinase (9). Besides AP-1, the inappropriate activation of other transcription factors such as NF-κB, C/EBP and CREB are also known to regulate cox-2 transcription (13), Therefore, we have extended our study to examine the effect of celecoxib on the activation of other transcription factors, and particularly NF-κB and C/EBP, in TPA-stimulated mouse skin in vivo. While celcecoxib has been shown to suppress NF-κB activation in several cell lines, our study shows that TPA-induced NF-κB activation in mouse skin remained unaffected by pretreatment with celecoxib.
The transcription factor C/EBP consists of three major isoforms: C/EBPα, C/EBPβ, and C/EBPδ, and these play a regulatory role in the induction of COX-2 by bacterial lipopolysacharides (LPS) or TPA (24). Therefore, we examined the effect of celecoxib on the TPA-induced activation of C/EBP in mouse skin. Our results of inducing C/EBP DNA binding and the elevated nuclear expression of C/EBPδ in TPA-treated mouse skin correlate with those of the previous studies that demonstrated an elevation of the nuclear levels of C/EBPδ, but not C/EBPα or C/EBPβ, in mouse skin papillomas (13) and LPS-stimulated murine macrophages (25). In addition to our previous study that reported on inhibition of AP-1 signaling as a molecular basis for the chemoprevention by celecoxib (9), the present study indicates that C/EBP is another potential target of celecoxib in achieving its antitumor promoting effects in mouse skin in vivo. It has been demonstrated that celecoxib inhibited the phosphorylation as well as catalytic activity of p38 MAP kinase, which regulates activation of such transcription factors as AP-1 and NF-κB in mouse skin (9). It would be quite interesting to conduct further research to explore the role of p38 MAP kinase for TPA-induced C/EBP activation in mouse skin.
CONCLUSIONS
In this study, topically applied celecoxib inhibited mouse skin tumor promotion. The anti-tumor promotional activity of this anti-inflammatory drug appears to be attributed to its suppression of COX-2 expression induced by the tumor promoter, TPA. While our previous work addressed the role of AP-1 in TPA-induced COX-2 expression in mouse skin, the present study suggests C/EBP as another potential transcription factor that may regulate COX-2 induction and can hence be a potential target of celecoxib.


 XML Download
XML Download