

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
1) Epstein-Barr virus: from discovery to group 1 carcinogen
Beginning in the 1940s, Denis Burkitt, a British missionary surgeon, observed and treated children in East Africa with previously undescribed extranodal lymphomas. In 1958, Burkitt first described an unusual undifferentiated non-Hodgkin's lymphoma (referred to as Burkitt's lymphoma), raising the spector of an infection etiology (1). In 1964, Epstein, Achong and Barr succeeded in establishing culture cell lines from samples of freshly excised tumor biopsies and finally demonstrated herpesvirus-like particles in the cultured cells using electron microscopy, and designated as Epstein-Barr virus (EBV) (2). EBV was shown to be the etiologic agent of heterophile-positive infectious mononucleosis in 1968 (3).
The EBV is a ubiquitous human herpesvirus which establishes a life-long persistent infection of B lymphocytes in over 90% of the human adult population (4). While in the vast majority this persistent EBV infection remains asymptomatic, a small proportion of individuals develop virus-associated tumors. Since the discovery of EBV in Burkitt's lymphoma in 1964, EBV has moved from being a bit-part player in the story of an obscure Burkitt's lymphoma to its present leading role as the prime example of a human tumor virus that is etiologically linked to an unexpectedly diverse range of malignancies. In addition to Burkitt's lymphoma, the list of EBV-associated tumors includes nasopharyngeal carcinoma (5), Hodgkin's disease (6), post-transplant lymphoproliferative disorders, T-cell non-Hodgkin lymphomas (7), gastric carcinomas (8), possibly breast (9,10) and hepatocellular carcinomas (11), and smooth muscle cell-derived tumors in immunodeficient individuals (Table 1) (4,12). These developments have greatly stimulated research into EBV-associated tumors. In 1997, EBV was classified as a group 1 carcinogen by the International Agency for Research on Cancer (4).
EBV-induced malignancy is unique in three respects. First, EBV viral genes involved in oncogenesis perform the same functions in normal latent virus infection. Second, EBV viral genes important for oncogenesis are normally expressed in latent infection. Third, EBV viral genes associated with oncogenesis are part of normal life cycle of the virus (12). Meanwhile, the idea of "hit-and-run" role in gammaherpes virus oncogenesis has led to many late-night discussions at scientific meetings (13). Staratschek-Jox and colleagues applied an in situ hybridization technique to apparently EBV-negative tumors to Hodgkin's disease in search of fragments of the viral genome (14). The methods applied are state of the art, and the answer appears to be clear. In contrast to the report of viral DNA fragments in sporadic Burkitt's lymphoma, no fragments of the viral genome were detected in the cases of Hodgkin's they studied. If the virus is involved in the pathogenesis of these EBV-negative cases, it seems to have made a clean escape, not leaving behind any DNA evidence for the lab to investigate further. Studies of Burkitt's lymphoma, nasopharyngeal carcinoma, and Kaposi's sarcoma cell lines suggest that such clean escapes are possible; thus, a "hit-and-run" role for EBV or KSHV in many diseases cannot be excluded. The challenge now is to devise investigative strategies that might lead to the conclusive identification of hit-and-run perpetrators or to exclude them definitively. The difficulty in devising such strategies has for the most part stopped investigators in the field from discussing the possibility of gammaherpes virus hit-and-run oncogenesis in print. Developing strategies to prove or exclude hit-and-run oncogenesis associated with episomal loss remains an interesting challenge.
2) EBV biology: lytic replication state and latent state
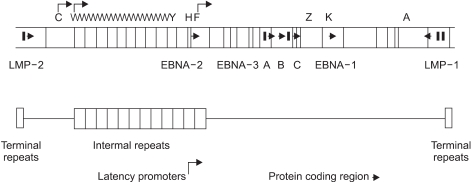
EBV is a member of lymphocryptovirus genus of gamma herpesvirus family. The EBV genome is a linear, double stranded, 184-kbp DNA (12). EBV has both lytic replication state and latent state as other kinds of herpesvirus do. In lytic replication, viral geneme maintains a linear form, and contains potentially over 100 open reading fragmes. The nomenclature of these open reading frames is based on a Bam HI restriction map of the viral genome (Fig. 1) (12,15).
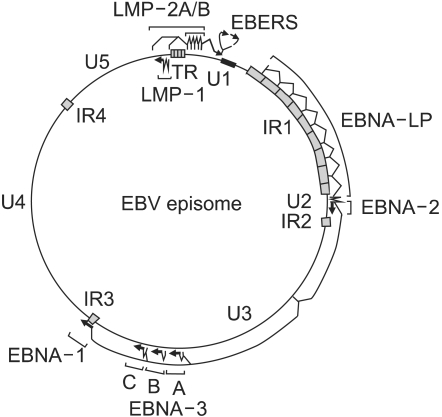
In infected cells, the EBV genome enters the nucleus, where it forms a circular episome (Fig. 2) (12). Episome formation is mediated by 0.5 kb terminal repetitive sequences located at either end of the linear molecule. Fusion of these sequences results in terminal repetitive regions with variable numbers of repeats (16). It is believed that individual infection events lead to episomes which differ in their number of repeat of terminal repetitive region; i.e. episomes within a single cell show the same number of repeats. Thus, analysis of the terminal repetitive region by Southern blot hybridization can provide evidence regarding the clonality of the viral genome (16).
In EBV-infected cells, virus replication with production of infectious virus is a rare event. Typically, EBV establishes a latent infection (4). The latent infection of EBV is characterized by the expression of a limited set of viral genes, the so-called latent genes, including two types of non-translated RNA (EBV-encoded nuclear RNAs; EBER1, EBER2), six EBV-encoded nuclear agents (EBNA1, 2, 3A, 3B, 3C, -LP), and three latent membrane proteins (LMP1, 2A, 2B), among the nearly 100 viral genes that are expressed during replication infection of EBV (12). ZEBRA (BZLF1) expression is inhibited in latent state, but certain stimuli can induce ZEBRA synthesis, and then make go to lytic replication state (17). Therefore, ZEBRA seems to play a role as an initiation of lytic replication state (4,17).
3) EBV entry into cells
EBV preferentially infects B lymphocytes through the binding of the major viral envelope glycoprotein gp350 to the CD21 receptor on the surface of B cells (18), and through the binding of a second glycoprotein, gp42, to human leukocyte antigen (HLA) class II molecules as a co-receptor (19). Infection of other cell types, principally epithelial cells, is much less efficient and occurs through separate, as yet poorly defined, pathways (19).
4) General course of EBV infection
EBV infects over 90% of the human adult population world-wide and, after primary infection, the individual remains a lifelong carrier. The oropharynx is the primary site of infection and is believed to be the site for virus replication (20). Primary EBV infection occurs usually in childhood and then is asymptomatic in most cases (21). In most industrialized countries, primary infection is delayed into aclolescence or early adulthood and then may cause a self-limiting lymphoproliferative disorder, infectious mononucleosis (3). Early in primary infection, EBV infected B cells can be found in large numbers in peripheral blood and tissues. In people with normal immune response, the number of latently infected B cells in the peripheral blood falls to approximately one in 105~106 during the months after primary EBV infection, a pattern that is associated with the alleviation of symptoms (22).
5) Immune defense to EBV and immune evasion of EBV
The presence of EBV in epithelial cells and B lymphocytes provokes an intense immune response consisting of antibodies to a large variety of viral antigens. In people with normal immune response, cells expressing EBV-encoded nuclear agents (EBNAs) and latent membrane proteins (LMPs) engender EBV-specific, major histocompatibility complex (MHC) class I-restricted, cytotoxic CD8+T-cell responses (12). Other defense mechanisms include neutralizing antibodies, cytokines such as interferons, natural killer cells, and antibody-dependent-mediated cytotoxicity (12,23). The EBNAs in particular, except for EBNA-1, have multiple epitopes that are recognized in the context of common class I determinants. The EBNAs and LMP1 also induce the expression of adhesion molecules, rendering the cell susceptible to T-cell adherence and cytocidal effects. As a consequence of immune responses by normal people to primary EBV infection, the number of proliferating virus-infected B lymphocytes in the peripheral blood rapidly declines to a level of one infected B lymphocyte in 105 or 106. However, cytotoxic T lymphocytes specific for epitopes from five of the EBNAs and the two LMPs persist forever, indicating that cells expressing the EBNAs and LMPs are at least intermittently present in the normal host (12).
Despite potent immune effector responses, the ability of EBV to persist indicates that the virus has evolved strategies to elude the immune system. EBV encodes a cytokine and a cytokine receptor that may be important for modulating the immune system to allow persistent infection. The EBV BARF1 protein functions as a soluble receptor for colony-stimulating factor (CSF)-1. Since CSF-1 normally enhances the expression of IFN-alpha by monocytes, BARF1 protein may function as a decoy receptor to block the activation of the cytokine (24). EBNA-1 has been shown to block its own degradation by proteosomes in infected cells (25). Since viral proteins are normally broken down by proteosomes to peptides for presentation to CTL, the ability of EBNA-1 to inhibit its degradation may allow the protein to avoid triggering the activation of CTL. In addition, EBV can modulate the ubiquitin-proteasome system to manipulate the host immune response, promote viral replication and inhibit apoptosis (26).
6) EBV latency and viral oncogenesis
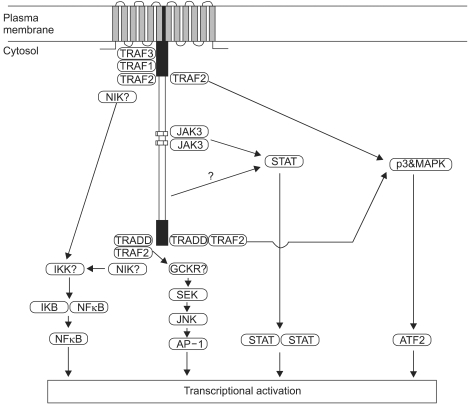
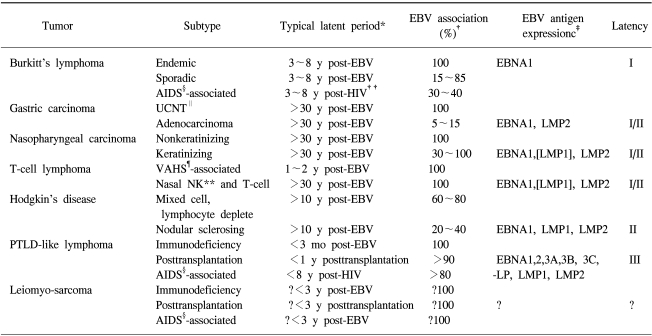
Three types of latency have been described, based on the variable expression of the latent gene products (Table 2) (4). The fact of that the patterns of viral latent protein expression in human malignancy are variable suggests that the contribution of EBV to different carcinogenic process may also vary. The latent genes are implicated in the process of transformation. In particular, LMP1 is the only EBV protein with recognized oncogenic activity. LMP1 can transform Rat-1 (fibroblast cell line) oncogenically (27) and transform epithelial cells morphologically (28,29). In addition, LMP1 transgenic mice develop hyperproliferation and lymphomas (30,31). LMP1 is established as a viral oncogene in EBV-associated lymphoma or nasopharyngeal carcinoma. It has been known about at least four signalling pathways by LMP1; namely nuclear factor kB (NF-kB), c-Jun N-terminal kinase (JNK)-AP-1, p38/MAPK (mitogen activated protein kinase), and Janus kinase (JAK)-STAT (signal transducers and activators of transcrption), are implicated in the function of LMP1 (Fig. 3) (12,32).
7) EBV detection method
The method used to detect the presence of EBV infection may vary between studies, potentially giving rise to variations in detection rate of the virus within different disease groups. The use of PCR to detect EBV has the obvious benefits of ease sensitivity. However, this exquisite sensitivity increases the likelihood of detecting EBV within non-malignant cells such as lymphocytes adjacent to tumor cells (33,34).
Although the detection of EBV genomes within infected cells can be accomplished with DNA in situ hybridization using Bam HI W repeats as a target, such studies have been criticized because of lack of sensitivity and poor signal to noise ratio.
The development of an in situ hybridization for the abundantly expressed EBERs provided a sensitive method for the detection of latent EBV infection in clinical tissues, including routinely processed histological material (35). Because EBERs are believed to be expressed in all forms of viral latency, in addition, EBERs exist abundantly, 106~107 copies per an infected cell, EBER in situ hybridization provides a consistent marker of latent infection and, perhaps because of the abundance of the EBERs, relatively short hybridization times usually suffice, proving a technique that can be completed in less than 24 horus (34,36~41). Therefore, that EBER in situ hybridization has been used extensively in studies demonstrating the association of EBV with a variety of disorders.
A range of monoclonal antibodies directed against latent EBV proteins, such as (LMP1, LMP2, EBNA1 and EBNA2 (42) has been developed.
EBV ASSOCIATED GASTRIC CARCINOMA
Gastric carcinoma represents the most significant EBV associated carcinoma in Korea, where gastric carcinoma is the most common malignancy and the second leading cause of death due to malignancy (43).
1) EBV: oncogenic virus in gastric carcinoma
EBV infected cells can induce specific genetic change, and initiate malignant transformation (44). Moreover, EBV is requisite to maintain malignant phenotype (45).
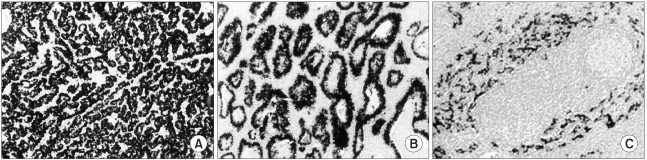
The following evidence supports that EBV is the oncogenic virus (not passenger virus) in patients with EBV-positive gastric carcinoma; Firstly, in-situ hybridization reveals the uniform presence of EBV infection in all carcinoma cells, but not in normal epithelium (34,37~41,46) (Fig. 4). Secondly, EBV DNA in carcinoma cells has been found to be monoclonal by Southern blot hybridization of the EBV terminal repeat fragment (46,47). Finally, there is serological evidence of high antiviral titers, especially EBV viral-capsid antigen IgA and EBV early antigen R component IgG, before diagnosis of the EBV-positive gastric carcinoma (48).
2) EBV infection rate in gastric carcinoma
Almost exactly fifteen years are going by after the first publication on EBV detection in three cases of gastric carcinomas (8). Now, it is well known that 2~16% of gastric carcinomas throughout the world reveals monoclonal proliferations of EBV infected carcinoma cells (49~51). EBV associated gastric carcinoma has been described in different populations from low-incidence areas, such as Western Europe and the United States, to high-risk countries, such as Korean and Japan (4). Although EBV has been detected in only a small proportion of gastric carcinomas, they are so common as to make EBV-positive gastric carcinoma a more significant health problem in terms of absolute case numbers than, for example, EBV-associated lymphoma. Gastric carcinoma is common among human malignancy, and the world wide occurrence of EBV positive gastric carcinoma is estimated at more than 75,000 cases/year (50,52). In particular, gastric carcinoma is the most common malignancy in Korea (43), and it was reported that 5.6% (38) or 13% (53) of Korean gastric carcinoma was associated with EBV. Consequently, gastric carcinoma represents the most significant EBV-positive malignancy in Korea.
The carcinogenic roles of EBV may be different in terms of each subset of gastric cancer. In other words, EBV detection rate is high in some subsets of gastric carcinomas, and is low in other subsets. Gastric carcinoma with lymphoid stroma of gastric carcinomas (GCLS) (Fig. 2) is 1.6% (54) or 3.7% (55) of the total gastric carcinomas and shows more favorable prognosis as compared with conventional adenocarcinoma (56). The EBV-positive rate in GCLS is ranged from 71% (57) to 84.6% (55). EBV infection rate in Korean GCLS is 67% (34). Moreover, lymphoepithelioma-like carcinoma is microscopically undifferentiated cancer without gland formation, as a subset of GCLS. The lymphoepithelioma-like carcinomas show strikingly high EBV infection rate regardless of geographic origin, ranging from 87.5% (50) to 93%. (52) Gastric remnant cancer after a primary gastrectomy for benign gastric disease, shows statistically higher EBV infection rate than in conventional gastric carcinomas, 27.1% (58), which is similar to 29% in Korean gastric remnant cancer (37). Bile reflux theory is suggested as a reason for high EBV infection rate in gastric remnant cancer. Meanwhile, metachronous cancer (so-called another type of gastric remnant cancer occurring after 5 years of subtotal gastrectomy for gastric cancer) shows 8% of EBV infection rate (37). Besides, gastric cancer of young patients ≤30 years old) or synchronous multiple cancer without accompanying adenoma show statistically significant higher EBV infection rate than the conventional gastric carcinomas (40).
3) Clinicopathologic characteristics of EBV associated gastric carcinoma
The EBV-positive gastric carcinomas tended to have lymphoid stroma. They were mostly of the poorly differentiated type, diffuse according to Lauren's classification, negative for p53 immunoexpression (possibly, negative p53 mutation), more prevalent in male patients and predominantly in the proximal stomach, particularly in the gastric cardia (38,44). It is also interesting that EBV asoociated gastric arcinoma in its intramucosal stage is likely to exhibit a specific histologic pattern; Abortive tubular structures with branching-anastomosing features occupy the middle of the mucosa without destroying the mucosal architecture, so-called lacy pattern (59).
The clinical outcome of EBV-associated gastric carcinomas was reported upon various analytical approaches. The prognosis of patients with advanced EBV-positive gastric carcinomas was not significantly different that of patients with EBV-negative carcinomas (38), while EBV-positive gastric carcinomas showed favorable outcome when metastatic carcinomas were evaluated (55). Recently, we reported that the hierarchical clustering of EBV-positive carcinomas according to tumor suppressor protein expression profile was considered to constitute a significant prognostic factor (60).
4) EBV specific immunity in patients with EBV associated gastric carcinoma
EBNA-1 and BARF1 are expressed in EBV-associated gastric carcinoma. Recently, BARF1 has been suggested as viral oncogene in EBV-associated gastric carcinomas. Patients with EBV positive gastric carcinoma have high IgG antibody titers against EBV capsid antigens (VCAs) and early antigens (EAs). IgA antibody against VCAs is detected in about 60% of cases, (Fukayama) but its diagnostic value is limited because the titers are much lower than those in nasopharyngeal carcinoma. There is evidence of high antiviral titers before the diagnosis of EBV positive gastric carcinoma (48).
EBV associated gastric carcinomas of the accompany lymphocyte infiltration. These tumor infiltrating lymphocytes are predominantly human leukocyte antigen (HLA) class I restricted CD8 positive cytotoxic T lymphocytes.
5) Controversy and peculiarity in EBV associated gastric carcinoma
There are three controversial points in EBV associated gastric carcinoma; viral oncogene, EBV entry mode into cell and EBV infection timing.
The viral oncogene in EBV-positive gastric carcinomas has not been yet established. The LMP1 is a classical viral oncogene in EBV-positive malignant lymphoma or nasopharyngeal cancer, but LMP1 is not expressed in EBV-positive gastric carcinomas (12,61,62). EBV exists in latent state in EBV-positive gastric carcinomas, and expresses EBV latent proteins such as EBNA1, EBERs and LMP2, and some lytic proteins (12,61,62). Several papers indicate that BARF1 might be a viral oncogene in several cell lines (63,64), and BARF1 induced tumor formation has been established in SCID mice (65). Moreover, BARF1 gene transcription was detected in nine out of ten EBV-carrying gastric adenocarcinomas (66). However, it is not known about molecular pathway of BARF1-induced oncogenic pathway, as well as there is no direct evidence of BARF1-induced transformation in gastric epitherial cell.
For the EBV entry mode into cells, three pathways have been suggested; CD21 receptor induced entry (67), EBV-specific IgA mediated entry (68), and cell fusion between EBV-carrying lymphocytes and epithelial cells (69). CD 21 receptor is not generally demonstrated in gastric epithelial cells, although epithelial cells of oropharynx, uterine cervix and salivary gland have CD21 receptor. Recently, we suggested that EBV carrying lymphocytes is a tissue reservoir of EBV in gastrointestinal tract, and the chances for epithelial cells to be exposed to the EBV are similar in the gastrointestinal tract regardless of site, and then may transfer the EBV to the epithelial cells by means such as cell fusion or secretory component-mediated IgA transport (33).
The timing of EBV-infection into gastric epithelium may be believed to occur in the earliest phase of its carcinogenesis, because of the results from EBER in situ hybridization and Southern blot hybridization of the EBV terminal repeat fragment. In all EBV-positive carcinomas, EBV has been detected in virtually all tumor cells using EBER in situ hybridization, and Southern blot studies have demonstrated clonal EBV episomes (16). This indicates that EBV infection in virus-associated carcinomas occurs before expansion of the malignancy cell clones. But, EBV infection probably occurs relatively late (after the adenoma stage) in the adenoma-carcinoma sequence, although EBV incorporation is rarely associated with the gastric adenoma-carcinoma sequence, moreover, the adenoma-carcinoma sequence seldom occurs in the stomach (39).
6) Molecular pathology in EBV associated gastric carcinoma
The exact mechanism by which EBV contributes to the carcinogenesis of the gastric mucosa remains unknown. Here are some facts of molecular events occurring in EBV associated gastric carcinomas. As to tumor-associated proteins including cell cycle regulators and apoptosis-related proteins, EBV associated gastric carcinomas show frequent loss of p16, smad4, FHIT and KAI-1. The p16 loss is predominantly caused by p16 promotor methylation, which occurs frequently in EBV (+) gastric cancer (70,71). There is negative association between EBV infection and the expression of MUC1, MUC2, MUC5AC, CEA, c-erbB2, smad7 and p53 (60). The p27, p16, cyclin D1 and NF-κB may be associated with oncogenesis in EBV-positive gastric carcinomas. EBV-positive gastric carcinomas show wild-type p53 stabilization and rare bcl-2 involvement. The characteristic expression of proteins may relate to both EBV and tissue type (41).
EBV associated gastric carcinomas belong to CpG island methylator phenotype-positive and suggests that aberrant methylation may be an important carcinogenic mechanism (71). CpG island methylator phenotype is characterized by widespread hypermethylation of CpG islands over the genome, and frequently show p16 promoter hypermethylation.
The information available regarding the relationships between EBV and MSI in carcinogenesis is both controversial and limited (72~74). In general, the relationship between MSI and viruses having human cell transforming properties has been little explored in human cancer. We evaluated Epstein-Barr virus (EBV) and MSI in various subsets of gastric carcinoma cases. None of the total 549 gastric carcinomas demonstrated both EBV positivity and MSI positivity. Furthermore, the EBV-positive and the MSI-positive cases showed a mutually negative association in all subsets of gastric cancer. Therefore, the carcinogenic roles of EBV and MSI may be different in terms of each subset of gastric cancer. EBV and MSI may contribute to functionally equivalent pathways in gastric carcinogenesis (40).
EBNA-1 may bring USP7 (HAUSP: herpesvirus associated ubiquitin-specific protease) to OriP where it could act on specific cellular substrates. It is also possible that binding to EBNA-1 may interfere with the physiologic function of USP7. USP7 was shown to bind and deubiquitinate p53, resulting in p53 stabilization and p53-dependent growth arrest and apoptosis (75). By sequestering USP7, EBNA-1 may destabilize p53 with important consequences for both B-cell immortalization and the development of EBV-associated tumors (76).
EBV-TARGETED THERAPEUTIC APPROACHES
Given the significant burden of EBV-associated tumors worldwide, an important priority is to design novel therapies that specifically target viral proteins or otherwise exploit the presence of the virus in malignant cells. Currently, EBV-targeted therapies are not made a trial in EBV-positive gastric carcinomas, although it has been attempting in another EBV-positive malignancy. EBV-based therapies are currently being developed for the treatment of EBV-positive malignancies. There are four potential strategies; inhibition of EBV transforming properties, loss of the EBV episome, purposeful induction of lytic EBV infection, and enhancing the host immune response to viral proteins.
1) Inhibition of EBV transforming properties
The oncogenic phenotype of some EBV-associated tumors can be reversed when EBV proteins responsible for transformation (such as LMP-1. EBNA-2, EBNA-3a and EBNA-3c) is inhibited (77). Recently, small interfering RNA (RNAi) technology has been demonstrated its highly effective selective gene inhibition. Expression of short (15~30 bp) double-stranded RNA sequences homologous to the target gene can lead to degradation of the target mRNA. The RNAi technology has been used to modulate viral expression in vitro of HIV (78), human papillomavirus (79) and so on. Therefore, RNAi technology is likely to be successful in the field of EBV. Meanwhile, the efficient delivery methods in vivo to tumor cells is a basic requisite for clinical use, and it has not yet been developed.
In model systems, LMP1 effector function has been targeted indirectly, by the genetic or pharmacological interception of its downstream effects on NF-kappaB (80). More recently, lymphoblastoid cell line growth in vitro has been impaired by blocking the transactivating function of EBNA2 using a short-peptide mimic of the RBP-Jk-interaction domain of the viral protein (81). LMP1 induced NF-kappaB activation plays a role in EBV-induced oncogenic property (12), and inhibition of NF-kappaB results in spontaneous apoptosis of lymphblastoid cell lines (80).
2) Loss of the EBV episome
Hydroxyurea treatment can make a loss of EBV episome (82), although the exact mechanism has not yet been defined. EBV episomes are thought to contain only one major ORC (origin of replication complexes) to regulate initiation of DNA synthesis. The in vitro inhibition of ORC formation may be useful (83), although in vivo data are not enough.
3) Purposeful induction of lytic EBV infection
Although a specific, nontoxic strategy for efficiently inducing lytic EBV gene transcription in tumor cells has yet to be developed, almost all agents used to activate lytic EBV infection in vitro are also capable of inducing cellular apoptosis (84). A number of including gamma irradiation and chemotherapy have the additional property of inducing lytic viral gene expression in a portion (up to 25%) of the tumor cells (85). EBV appears to be capable of recognizing a host cell environment indicative of cellular stress, and uses these signals to convert to the lytic form of infection, and escape a potentially dying host cells. Once the conventional treatment for EBV-positive tumors induces a portion of the tumor cells to convert to the lytic EBV infection, antiviral drug, ganciclovir, might augment the therapeutic effect of chemotherapy or irradiation (86).
(a) Expression of EBV immediate early gene products
The switch from the latent to lytic form of EBV infection is mediated by the two viral immediate-early proteins, BZLF1 and BRLF1. Upon transfer BZLF1 into EBV-positive lymphoma cell lines, the latent virus switched to a lytic cycle, which resulted in lysis of the neoplastic cells (87).
(b) Modification of chromatin or DNA methylation
In cells containing tightly latent EBV infection, the viral immediate-early promoter DNA is often methylated (88), and the chromatin surrounding the immediate-early promoters is in the unacetylated (inactive) form (89). Histone acetylation inducing agents (85) or reverse DNA methylation inducing agents (90) can in vitro lead to lytic form of EBV infection in subset of cells in Burkitt's cell lines. Although histone acetylation inducing agent, such as butyrate, and demethylating agent like 5-azacytidine induce lytic infection, treatment of mice with either butyrate or 5-azacytidine could not induce effectively the lytic infection of EBV in lymphoblastoid cell line-derived tumors (85).
4) Enhancing the host immune response to viral proteins
EBV-specific cytotoxic T lymphocytes therapy appears to be most effective for treating post-transplant lymphomas (91,92). EBV-specific cytotoxic T lymphocytes strategy was highly effective, both as a therapy for the treatment of existing disease, and in prophylaxis (93). EBNA-2, EBNA-3a, -3b and-3c, and LMP-2 contain the immunodominant epitopes for latent EBV proteins in normal cytotoxic T lymphocytes responses (94).
Clinical trials of EBV-specific cytotoxic T lymphocytes therapy for type II latency tumors such as Hodgkin's lymphoma and nasopharyngeal carcinoma, in which viral gene expression is restricted to EBNA-1, LMP-1, LMP-2 and BARF0, represent just a first step. (95). Strategies are not being developed either to generate T-cell preparations for transfer that are enriched in CD8+ and possibly CD4+; reactivities to available subdominant targets (such as LMP2A and EBNA1) (92,96), or to immunize the patient with appropriate antigenic constructs to boost these particular responses in vivo (97). In fact, tumors can develop in hosts with apparently intact cytotoxic T lymphocytes function, and this suggests that paracrine effects such as TGF-beta and IL-10 may restrict the function of EBV-specific cytotoxic T lymphocytes. In order to resist such local tumor immunosuppressive effects, attempts to modify EBV-specific cytotoxic T lymphocytes ex vivo was made (98).
EBV-TARGETED VACCINATION
In 1982, Kieff and his research team launched an effort to develop an EBV vaccine. Tests of its safety and efficacy began three years ago in several hundred volunteers, including a small number of patients (99). Theoretically, EBV envelope glycoproteins, gp340, gp350 and gp85 have been suggested as the target of EBV vaccine (100~102).
CONCLUSIONS
The picture which has emerged after over 40 years of EBV research is complex. Currently, the precise mechanisms by which EBV transforms B lymphocytes are only being elucidated. In EBV-positive lymphoma, there are several LMP1-induced cellular signaling pathways including NF-kappaB, JNK, p38, STAT. Recently, novel therapies of viral targeted strategy and the project of vaccination have been developing, especially, in lymphoma.
EBV-associated gastric carcinoma is a unique type of gastric carcinoma that is tagged by clonal EBV, and might become a relatively more important gastric carcinoma if the frequency of other risk factors seems to feel declining. However, only a handful of knowledge exists in EBV-positive gastric carcinoma.

 XML Download
XML Download