

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Prostate cancer is one of the most commonly diagnosed malignancies in males. Mifepristone, a nonsteroidal antiprogestin, has been widely used to treat prostate cancer, through the activation of apoptotic pathways (1,2). However, the process of the initiation of caspase activation remains unknown.
For the initiation of apoptosis, the mitochondria play a crucial role by releasing apoptogenic factors, such as cytochrome c, from the mitochondrial intermembrane space into the cytoplasm (3). Cytochrome c triggers a cascade of caspase activation events, leading to apoptosis (4). The release of cytochrome c from the mitochondria is affected by several inducers, including reactive oxygen species (ROS). It is well known that the release of cytochrome c is induced by H2O2. Nuclear translocation of p53 is also known to be induced by H2O2, and p53-induced apoptosis requires the generation of ROS (5). p53 protein can be considered as one of the oxidative stress response transcription factors, and ROS is believed to be involved in the activation of p53 (6). p53 can be considered as one of the oxidative stress response transcription factors.
The mechanism of apoptosis is executed with a cascade of sequential caspase activations. Caspase-3, -6 and -7 are especially responsible for the execution of apoptosis, byoperating at the downstream end of the DNA repair enzyme poly (ADP-ribose) polymerase (PARP), whose cleavage is essential for the induction of apoptosis (7,8).
Estrogen has been proved to be a powerful hydroxyl radical scavenger, which blocks the Fenton-reaction-ROS production (9). Therefore, mifepristone could block prostate cancer cell growth more efficiently when combined with tamoxifen, an antiestrogen, which is used in breast cancer treatment.
Polyamines are essential for normal cellular growth (10), and are known to block the generation of ROS and protect DNA from ROS damage (11,12). Interference with the biosynthesis and function of polyamines are an attractive anticancer chemotherapeutic strategy for prostate cancer. At present, there are very limited data on the usefulness of polyamine analogs against prostate cancer (13).
Thus, the purpose of the present study was to investigate if mifepristone can induce apoptosis through the generation of ROS, by studying the caspase-3 activation, p53 expression and PARP cleavagein LNCaP human prostate cancer cells. Estrogen and polyamines,known as free radical scavengers, were used for further proof of the ROS-initiated apoptosis by mifepristone.
MATERIALS AND METHODS
1) Chemicals
The 11β-[4-Dimethylamino]phenyl-17β-hydroxy-17-[1-propynyl] estra-4,9-dien-3-one (Mifepristone; RU-486), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium (MTT), 17β-estradiol (E2), putrescine (tetracethylenediamine), spermidine (N-[3-aminopropyl]-1,4-butanediamine), spermine (N,N-bis[3-aminopropyl]-1,4-butanediamine), 2,7-dichlorofluorescein diacetate (DCFH-DA), 2,7-diamino-10-ethyl-9-phenyl-phenanthridinium bromide (ethidium bromide) and Dulbecco's modified Eagle's medium (DMEM, with L-glutamine and 1,000 mg/l Glucose) were purchased from Sigma Chemical Co (St. Louis, MO.). The fetal bovine serum (FBS) was purchased from GIBCO (New York, NY). The anti-PARP antibody (mouse IgG), antibody for Caspase-3 (rabbit IgG), anti-p53 antibody (goat IgG), secondary anti-mouse IgG, secondary anti-rabbit IgG and secondary anti-goat IgG were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA). All other chemicals were purchased from standard commercial sources.
2) Cell culture
The parental LNCaP prostate carcinoma cell line was obtained from the American Type Culture Collection (ATCC, Rockville, MD), and maintained in DMEM, containing phenol red, with 10,000 units/ml penicillin G, 10 mg/ml streptomycin and 7% heat-inactivate FBS, in a humidified atmosphere of 95% air; 5% CO2, at 37℃. The culture medium was changed every 2 or 3 days. Cells were harvested using trypsin-EDTA, and subcultured at weekly intervals. Prior to each experiment, cells were grown for 2 weeks, in phenol red-free DMEM, containing serum treated with dextran-coated charcoal (DCC), to remove the serum-derived estrogenic compounds.
All experiments were conducted in phenol red-free DMEM and DCC-treated serum. Proliferation of the LNCaP cells, under these conditions, was necessary to obtain the maximal sensitivity to estradiol, and to avoid the estrogenic effects of the phenol red.
3) Measurement of cell viability
The cell viability was determined by the MTT assay. Cells treated with mifepristone (MIF) for 4 days were used for the viability analysis. MIF was dissolved in 100% ethanol, and then diluted to 1,000 times its volume in the same culture medium to achieve desired concentrations. The MTT stock solution (5 mg/ml PBS), freshly prepared and filtered through a 0.2 µm filter prior to use, was added to each culture, and incubated for 3~4 hr. After incubation, the medium was removed, and the converted dye solubilized with dimethyl sulfoxide (DMSO) and ethanol (1:1). The absorbance of the converted dye was measured at a wavelength of 570 nm.
4) Assay for Reactive Oxygen Species (ROS)
The detection of the intracellular H2O2 production was based on the oxidation of the nonfluorescent substrate, 2",7"-dichlorofluorescein (DCFH), to the fluorescent product, 2",7"-dichlorohydroxyfluorescein (DCF). The cells were seeded at 1.5×103 cell/well, in 96-well plates, allowed to attach overnight, and then treated with MIF. The DCFH-DA, dissolved in ethanol, was diluted with phosphate buffer (PB), to a final concentration of 10 µM, applied to the cells and incubated for 2 hr, at 37℃. The fluorescence intensity was measured with a fluorescence plate reader (BIO-TEK Instruments, Inc., Winooski, VT), with excitation and emission wavelengths of 485 and 530 nm, respectively.
5) Western blotting
Cells were collected by centrifugation, at 4,000 rpm and 4℃ for 5 min. After washed twice with ice-cold PBS, the cells were incubated, on ice, for 60 min, in lysis buffer, containing 50 mM Tris-HCl (pH 7.6), 300 mM NaCl, 0.5% Triton X-100, 2 mM phenylmethylsulfonyl fluoride, 2 µg/ml aprotinin and 2 µg/ml leupeptin, for the extraction of the protein. The cell lysates were centrifuged at 13,000 rpm for 20 min, and the protein concentrations determined by the Bradford method, using bovine serum albumin as the standard (14).
For western blotting, a 60 µg aliquot of protein was separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis, in 5~20% gradient polyacrylamide gels, and transferred to nitrocellulose transfer membranes. The blotting was performed using the primary antibodies of antiPARP (1:1,000 dilution), anticaspase-3 (1:1,000 dilution) and antip53 (1:1,000 dilution), and then with the secondary antibodies of anti rabbit or anti mouse IgG antibody (1:1,000 dilution). The immuno-complexes were detected using an ECL (enhanced chemiluminescence) detection kit (Amersham, Piscataway, NJ).
RESULTS
1) Influence of MIF on cell proliferation
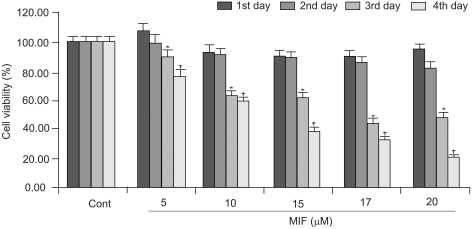
When the LNCaP cells were treated for 1~4 days, with 5~20 µM of MIF, their growth and viability were significantly decreased, in dose- and time-dependent manners (Fig. 1). The cell viability was uninfluenced by 2 days of MIF treatment, but after 3 days, the cell viability decreased significantly.
Cells treated with 17 µM MIF showed 90, 85, 44 and 33% viabilities, after 1, 2, 3 and 4 days of incubation, respectively, compared to the control. With 5, 10 15, 17 and 20 µM MIF treatment for 4 days, the proliferation of LNCaP cells were reduced to 77, 59, 39, 33 and 20%, respectively, compared to that of the control.
2) Effects of E2 and polyamines on MIF-induced cell death
The E2 and polyamines treatment blocked the effect of the MIF. Treatment with 17 µM MIF severely reduced the cell viability after both 2 and 4 days of incubation. However, the treatment with E2 or polyamines for 24 hr prior to the MIF treatment strongly inhibited the effect of the MIF on cell death (Fig. 2). The inhibitory effect against MIF was most prominent with the putrescine treatment.
3) MIF-induced ROS generation
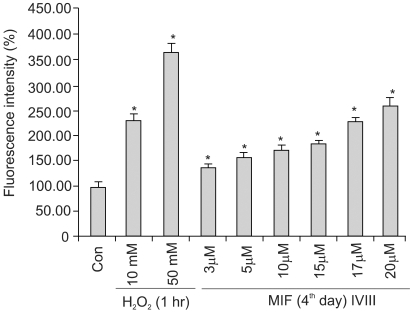
Fig. 3 shows the MIF-induced ROS generation to be dose dependent. With 3, 5, 10, 15, 17 and 20 µM MIF treatment for 4 days, the ROS production increased to 134, 159, 174, 189, 226 and 277%, respectively, compared to that of the control. When the cells were treated with higher concentrations, over 20 µM, the ROS generation did not significantly increased, and by no more than 3-fold that of the control. The ROS generation was increased in a time-dependent manner during the 4 days of MIF treatment (data not shown).
4) Influence of E2 and polyamines on MIF-induced ROS generation
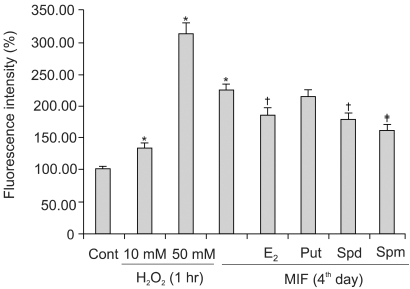
Fig. 4 shows the effects of the E2 and polyamines (PA) on the MIF-induced ROS production. The cells were treated with E2 and each of the PA for 24 h prior to the addition of 17 µM MIF. The pretreatment of the cells with 10 nM E2 resulted in a 58% inhibition of the MIF-induced ROS generation. In the pretreatment of the cells with each PA, 5 mM putrescine showed a minor inhibitory effect on the ROS generation, but spermidine and spermine, at 10 µM, exerted 55% p53 expression and 79% inhibition, respectively, of the ROS generation.
5) Influence of MIF in p53 expression
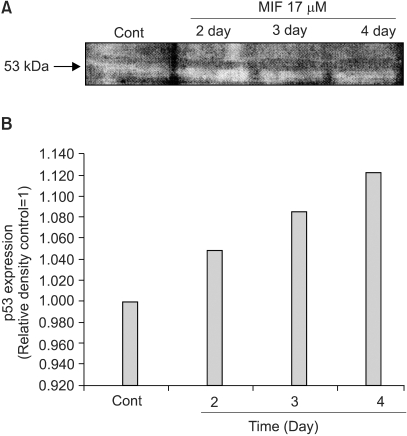
It has been suggested that p53-induced apoptosis is mediated through the generation of ROS. Nuclear translocation of p53 is also known to be induced by H2O2 (5). Fig. 5 shows the time course influence of MIF on the p53 protein level. LNCaP cells were treated with 17 µM MIF for 4 days. The results indicated that the p53 protein level was enhanced as the exposure time increased, from 2 to 4 days.
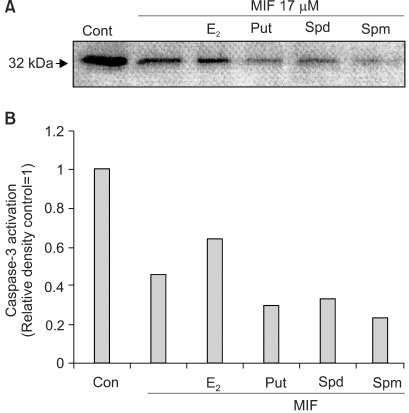
6) Effects of E2 and PA on MIF-induced caspase-3 activation
The activation of caspases is known to be involved, as a general mechanism, in the induction of apoptosis. The inactive form of caspase-3 is cleaved, and converted to an active form. When the effect of 17 µM MIF was evaluated on the activation of caspase-3, by measuring the cleavage of pro-caspase-3 (32 kDa), using Western blotting, the MIF-induced pro-caspase-3 cleavage increased in a time-dependent manner during the 4 days of incubation (data not shown). In the cells pretreated with E2, prior to the MIF treatment, the caspase-3 activation was significantly suppressed (Fig. 6). However, PA was unable to block the caspase-3 activation; rather caspase-3 was more activated in the presence of PA.
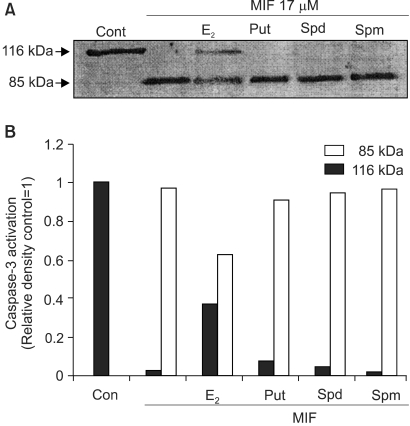
7) Effects of E2 and PA on MIF-induced PARP cleavage
PARP is a repair enzyme, which is cleaved and inactivated by caspase-3 and -7, and DNA fragmentation factor (DFF) (8). PARP cleavage, from a 116 kDa protein to an 85 kDa fragment, is considered as a hallmark of apoptosis. Treatment with 17 µM MIF to the LNCaP cells resulted in the cleavage of PARP. The cleavage of the PARP was observed to be time-dependent (data not shown), and that induced by the MIF was significantly inhibited by the E2, but the PA had no significant influence on the PARP cleavage (Fig. 7).
DISCUSSION
It has been reported that MIF increases DNA fragmentation and a bcl-2 down-regulation in androgen-sensitive LNCaP cells (15). The anti tumorigenic activity of MIF could be the result of the activation of the cell's suicidal mechanism; apoptosis. The present results clearly demonstrate the growth inhibition of MIF, through the apoptosis of the LNCaP human prostate carcinoma cell line. The growth inhibition was dose and time dependent. The activation of caspases, which is followed by the cleavage of PARP, is a critical step in the cell death induced by anticancer drugs. The present study provesthe caspase-3 activation and PARP cleavage in MIF-induced apoptosis.
The present results provide the first evidence indicating the induction of apoptosis through the MIF-induced ROS generation on LNCaP cells. The MIF-induced ROS generation resulted in caspase-3 activation. In addition, the present study also shows that MIF is able to induce the overexpression of p53 protein in LNCaP cells. It has shown that p53 can induce apoptosis through multistep processes, including the formation of ROS, the oxidative degradation of mitochondrial components and the transcriptional induction of p53-induced genes (16). The nuclear translocation of p53 could be induced by H2O2, and p53-induced apoptosis requires the generation of ROS (17). The above findings were demonstrated by the present results. The increased intracellular ROS levels and p53 expression were detected after the incubation of LNCaP cells with MIF. Wang and his colleagues (6) reported that the .OH radical is the key species among the ROS responsible for p53 activation. In the present experiment, the MIF-induced apoptosis was also clearly demonstrated by the cleavage of the DNA repair enzyme, PARP, whose cleavage is essential for apoptosis (7).
Androgens are essential for stimulating the growth and development of the prostate, but estrogens generally inhibits its growth. Therefore,estrogens have often been used in the treatment of prostate cancer. Apart from their effects on the hypothalamic-pituitary-testicular axis, there are also indications for the direct action of estrogens on tumors, via cytotoxic mechanisms and through the decreased formation of the terminal biologically active androgen, 5α-dihydrotestosterone, in tumor tissue (18). In the prostate, the action of testosterone is dependent on its conversion to dihydrotestosterone, and on E2, via 5alpha-reductase and aromatase, respectively. However, the influences of estrogens in prostate cancer are controversial. Most recently, it was reported that malignant changes to the prostate gland were dependent on both androgenic and estrogenic responses, and that neither hormone alone was sufficient to induce aberrant patterns of growth in a murine prostate (19). In a previous report, the co-treatment of MIF and tamoxifen was more effective at inducing apoptosis in prostate cancer cells than MIF alone. These findings indicate that estrogens interfere with apoptosis. The chemical structure of estrogen allows for the donation of an H+ ion to a peroxyl radical. This property allows estrogens to act as free radical scavengers. Estrogen may exert its effect by interfering early in, or during, the mid-propagation phase of lipid peroxidation, which is known to be most frequently induced by O2- and H2O2 (20). The present results clearly show that E2 reduced the ROS production level to 58% that of the untreated control in the MIF treated cells. E2 treatment also severely suppressed the caspase-3 activation and PARP cleavage. The previous reports that the combination of MIF and tamoxifen severely induced apoptosis in LNCaP cells support the roles of E2 found in the present experiment.
Naturally occurring polycationic polyamines are found in all eukaryotic cells. Spermine and spermidine are known to stabilize chromatin and nuclear enzymes, by forming complexes with organic polyanions. Natural polyamines have also been shown to inhibit lipid peroxidation (12). In the present study, PA, especially spermine, had a noticeable antioxidant effect on the MIF-induced ROS production. However, polyamine pretreatment could not prevent the MIF-induced caspase-3 activation, and PARP cleavage, indicating that ROS generation is not the sole initiator of the apoptotic mechanism.
CONCLUSIONS
MIF, an antiprogestin, induces apoptosis through the generation of ROS, the overexpression of p53, caspase-3 activation and PARP cleavage in LNCaP human prostate cancer cells. E2 prevents apoptosis, by working as a free-radical scavenger, and also decreases the MIF-induced caspase-3 activation and PARP cleavage in the apoptotic process. Although polyamines, especially spermine, prevented ROS generation, all three polyamines tested had no influence on the caspase-3 activation and PARP cleavage. This implies that estrogen is multifunctional, rather than working only as a free radical scavenger, in the process of apoptosis.

 XML Download
XML Download