

 PDF
PDF Citation
Citation Print
Print


Evidence for a p53-like gene was first published in 1997 (16) in which it was designated "p73" to honor the original member of the family discovered by Levine and Lane nearly two decades earlier (17,35). p73 was more than just a member of the family- the similarity of the DNA binding domains of p73 and p53 was such that p73 transactivation activity was readily followed using the standard promoter-reporter constructs developed for p53. Thus it was apparent that, at least under experimental conditions, p73 had the potential to regulate p53 target genes. Additionally, p73 overexpression triggered apoptosis in many established cells and therefore mimicked yet another feature of the vaunted tumor suppressor (15). The similarities did not end there, however, as in situ hybridization showed that the p73 gene was ensconced in the center of a region of chromosome 1 known to harbor one or more tumor suppressor genes involved in neuroblastoma, breast and prostate carcinoma, as well as melanoma (16,30). Therefore p73 not only looked like a tumor suppressor but was a candidate for one of the long sought tumor suppressors in the 1p36.3 locus lost in many human cancers.
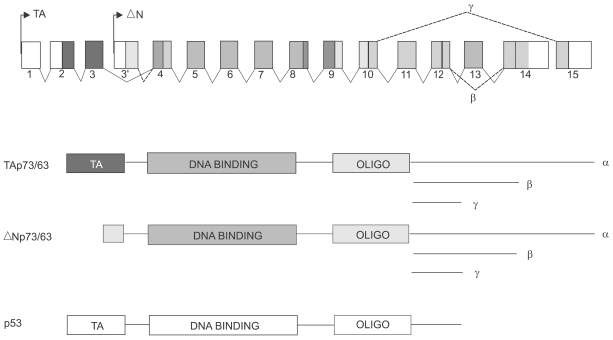
The discovery of the p63 gene came one the heels of the p73 description and was presented by at least five labs within a short time frame (2,21,26,28,31). Probably the most interesting revelation from the p63 cloning was that this gene, unlike p53, expressed transcripts off of two promoters and gave rise to multiple transactivating (TA) and N-truncated, potentially dominant negative (DN) isoforms (31)(Fig. 1). This gene structure was also found in p73 (34) such that it too produced both TA and DN isoforms. The significance of these findings was that these newly described genes might not only mimic p53 but potentially counteract p53 function as well. Additionally, the p63 gene was, unlike p73, not located in within a tumor suppressor locus but rather within a region of chromosome 3 that is in fact amplified in many cancers (11). Thus p63 location might implicate it more as an oncogene than as an obvious tumor suppressor. Regardless, the immensely complex structures of the p73 and p63 genes promised major challenges to understanding their functions with and besides regards to the p53 gene.
p73 as a Trigger for Apoptosis in Response to DNA Damage
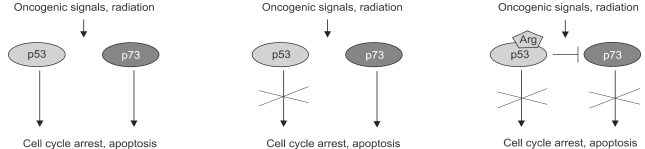
Some of the very early functional experiments done with the original TA isoform of p73 indicated that it could trigger apoptosis when expressed in cells in culture (15). In addition, this work showed that p73 could also cause cell cycle arrest through the transactivation of the p21 gene. Thus in every way p73 was acting, at least under experimental conditions, in a manner reminiscent of p53. From that initial point investigators examined the apoptosis generated in cancer cells that had mutant p53 (1,10,36,29). Cisplatin treatment of cells was linked to an enhanced steady state level of p73 and followed by increased apoptosis that was shown to require non-receceptor tyrosine kinase c-Abl. Parallel studies revealed that ionizing radiation could also influence p73 activity, this time by increasing the degree of phosphorylation of p73 by c-Abl. Thus multiple reports detailed apoptotic responses in which p73 was at the heart of an alternate path to cell death triggered by any number of cellular stresses that constitute the basic approaches to chemotherapy. The inevitable conclusion here was that p73 was acting somewhat as a backup for p53- it could carry on after p53 was wiped out. More ominous, however, was the very exciting finding that some mutant forms of p53 form strong interactions with p73 in a way not dependent on the oligomerization domain but more likely with the DNA binding domain of p73 (5). By this mechanism a single mutation in p53 could also inactivate p73 and therefore take out what could represent a parallel system of controls governing the response to cellular stresses in precancerous cells. Validation for this notion came from studies of p53 mutation patterns in squamous cell carcinoma (19). For some time investigators had noticed the preferential mutation and retention of one of two p53 polymorphisms at residue 72. Commonly an ARG or a PRO, the squamous cell tumors show a strong bias for the ARG allele. Remarkably, the ARG p53 allele is also the one that interacts most strongly with p73 therefore providing hints into the molecular mechanisms by which this bias originates (Fig. 2). This work also provides strong support for the importance of inactivating p73 in the process of tumorigenesis. The overall clinical significance of p73 status has been highlighted recently by the finding that p73 plays an important role in chemosensitivity of cancer cells (14). In this work it is shown that a large number of drugs used in chemotherapy induce p73 expression, and that the disruption of p73 expression, either through the use of dominant negative p73 constructs or siRNA enhances the chemoresistance of the tumor cells. Consistent with earlier studies, p53 mutants known to interact with p73 also enhances the resistance of cells to chemotherapy. These data not only support a role for p73 in cell death but also lend credence to the notion of gain of function alleles of p53. Together these findings underscore the ability of the p73 protein to mediate p53-like functions in lieu of p53. This quality, coupled with p73's stabilization in response to chemotherapy, suggests its eventual use in gene therapy trials for cancers.
p73 as a Critical Player in E2F1- and Activation-Induced Cell Death
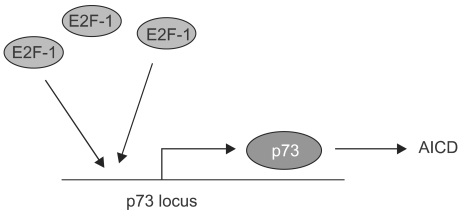
The E2F1 gene has come under considerable attention for its ability to promote cell death in response to cellular stress and has been generally attributed a role in tumor suppressor pathways (23). E2F1 can kill cells in both p53-dependent and independent ways. In a remarkable set of experiments, several laboratories managed to link the p53-independent means of cell death to p73 (13,18,27). The first set of experiments started with the ability of E2F1 to trigger cell death and then showed that E2F1 expression was sufficient to trigger the transactivation of the p73 gene and the induction of p73 protein (13,22,27). Significantly, the authors showed that in p53 mutant human cell lines and murine embryonic fibroblasts (MEFs) lacking p53, E2F1 expression also promotes apoptosis and does so in a way largely dependent on p73 (Fig. 3). Accompanying this work was another exciting study based on an earlier observation that E2F1 deletions in mice lead to increases in T cell number and splenomegally. Given that E2F1 can transactivate p73, the authors figured that p73 might mediate mature T cell apoptosis. A model for this form of T cell death is activation-induced T cell death (AICD) where T cells are overstimulated by activation of their T cell receptors. Interestingly, the authors not only showed that p73 is greatly induced by the activation of the TCR, but that p73 itself is essential for the cell death that follows this activation step (18). Thus p73 is shown to be important not only for pathways of cell death in chemotherapy but also for those that might underlie regulation of the immune response.
p73 and p63 Roles in Supporting p53 Function
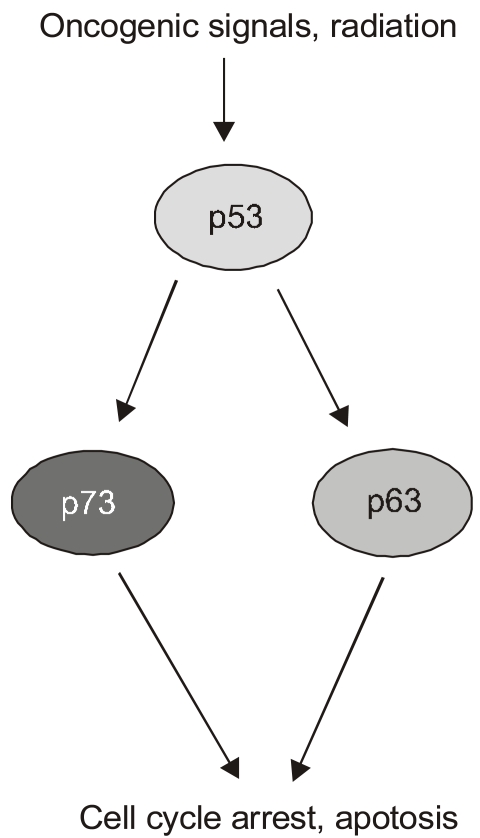
Most of the experiments put forth to argue for p73's and p63's roles as tumor suppression have yielded data consistent with that notion but suffer, to a greater or lesser extent, from nonphysiological design features. For instance, they usually involve p73 or some other factor that is overexpressed from exogenous promoters, a host cell from an established cell line derived from human tumors, and cellular stress programs only thought to mimic those facing cells in vivo. Therefore it was tremendously exciting to see the analysis of p73 and p63 function in genetically defined cells and mice (7)(Fig. 4). In particular, this study examined the roles of all of the p53 family members in the apoptotic response of MEFs to DNA damaging agents. As is standard in such analyses of p53-dependent cell death in MEFs, these cells were pre-programmed with the E1A oncogene that acts to sensitize the cells to apoptotic pathways. In this case, doxorubicin was used to introduce DNA damage in a process that kills a majority of the E1A MEFs over a period of 30 hours. Significantly, the absence of p53 abrogated much of this doxorubicin-induced cell death, indicating that neither p63 nor p73 was sufficient to assume this function in the absence of p53. However, further studies revealed a remarkable and unexpected property of the p53 homologs. The absence of either p73 or p63 resulted in a significant loss of p53-dependent cell death due to doxorubicin, though not nearly the effect of losing p53. Remarkably though, the loss of both p73 and p63 resulted in E1A-MEFs that were highly resistant to doxorubicin to a degree approximating that due to a genetic loss of p53. These experiments in E1A-MEFs were followed up with the analysis of apoptosis in fetal brain resulting from gamma-radiation (5 Gyr), another p53-dependent pathway of cell death. As expected, embryos lacking p53 showed very low levels of neuronal cell death due to this radiation. Again, however, the loss of p73 and p63 mimicked the effects of a p53-deficiency. The inevitable conclusion of this work was that the homologs were somehow required for p53-dependent cell death even though they had no ability to mediate doxorubicin- triggered cell death in the absence of p53. If these genes were not acting autonomously by paralleling the actions of p53 in transcriptional programs, how were they working? The authors examined the promoters of genes transactivated by p53 in response to DNA damage and noted that in wild type E1A MEF cells, both p53 and p63 could be seen at most of these sites by CHIP analysis. Intriguingly, in the E1A-MEFs lacking both p73 and p63, many of these genes, including PERP, Noxa, and Bax, showed no p53 binding to their regulatory DNA sequences. While the mechanistic significance of this finding is not obvious, one possibility is that p63 and p73 somehow recruit or stabilize the binding of p53 at promoters critical for the apoptotic response to DNA damage. The implications of these findings are immense with respect to the roles of p73 and p63 in tumor suppression. They suggest that neither one is essential for p53 function and yet together they are essential for p53 function. These data might also explain why neither of these genes is mutated in cancers. The possibility that both genes would be mutated before the p53 gene is altered would be unlikely and therefore there is a strong selection for p53 changes but not of the other two family members. Probably the major experimental liability of this work was the use of the E1A gene to potentiate the apoptotic response to doxorubicin. The ability of E1A to affect major cell cycle pathways in the cell is well established and therefore presents a cellular environment different from that of a wild type cell, although in ways we do not fully comprehend.
Absence of Tumors in p73-null Mice
Despite the groundswell of data supporting the tumor suppressor functions of p73 and p63, the mouse knockout models have been surprisingly uninformative on this issue. The p63 knockout mouse (32) lacks many epithelial tissues, including skin, due to the lack of underlying stem cells and therefore die soon after birth. Mice bearing conditionally disrupted p63 genes are in production that may permit the analysis of this gene in specific tissues. Regardless, determining the consequences of losing p63 on tumorigenesis is therefore not possible with this mouse model. The situation is much different for the p73 null mouse because some of these mice live to be 15 to 20 months and therefore can be followed for tumors (34). In fact the early mortality in these mice is not due to tumors but rather linked to malnutrition, hydrocephalus, or idiopathic gastrointestinal bleeding. In the mice that survive past the first thirty postnatal days, most live to advanced age. Nearly 100 of the p73-null mice have been analyzed for tumors at three to 12 months. Tumors are rare in these mice and appear at a rate similar to that seen in wild type mice. This has to be contrasted with the findings with either p53 homozygous or heterozygous mice, which show very high rates of tumor development (6). The bluntness of this observation is curious in light of the immense amount of data supporting p73's tumor suppressor function. Whereas this result could be explained and diffused by several arguments that would leave p73's role as a tumor suppressor intact, it is probably important to keep the mouse phenotype in view at these debates.
Oncogenic Functions of p73 and p63: the Evolutionary Viewpoint
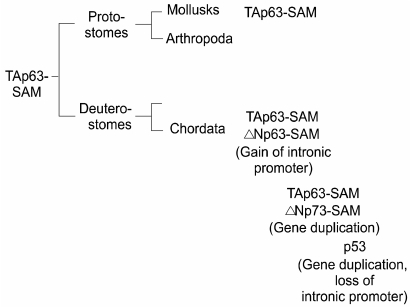
The discovery of the dominant-negative isoforms of p63 (31) and of p73 (34) set in motion an entirely new possibility that these genes could actually have functions that oppose those of p53. Indeed in transcriptional assays in cells ΔN-p63 and ΔN-p73 can neutralize p53 transcriptional activity by simple competition for binding sites on p53 promoter reporter constructs (34). The question becomes whether cancers can be initiated in some epigenetic manner by ΔN-isoforms expression without the need to mutate p53. This issue was initially raised by the mapping of the p63 gene to chromosome 3q27-a region known to be amplified in a wide range of cancers (11). Unfortunately the amplified region is large and contains many genes, including candidates for potential oncogenes such as a catalytic subunit of PI3 kinases (25). Regardless of the amplification issue, there is recent evidence that high levels of p63 expression, especially of the ΔN-isoforms, is a common feature of squamous cell carcinomas of the head and neck as well as other areas (8,12). What is lacking in this scenario is definitive evidence that p53 is suppressed by p63, especially in some epithelial cancers that derive from cells that express copious amounts of p63 such as skin and other squamous epithelial tissues. Experimentally this is difficult to obtain and hard to quantify if attempted. If such an interaction between these genes did exist, especially between ΔN-isoforms and p53, we might expect to see this reflected in the evolution of the family. Our analysis of the evolutionary relationships amongst the p53 family yielded several surprising results (33). The first is that the single p53-like gene in flies and worms was in fact more related to p63 than to p53 as argued (4,20), and that the original member of the family, widely distributed and conserved in many phyla, was in fact p63. The second is that in all phyla but Chordata, the only isoform of p63 is the transactivating "TA" version (Fig. 5). Thus in mollusks we see a highly conserved version of TA-p63 complete with a C-terminal SAM domain observed in human TA-p63 alpha, indicating that this was the original gene structure before the split between deuterostomes and protostomes (9). The immediate implication of this is that unlike p63, p53, as well as p73, are only seen in vertebrates. As important, the first time we see the dominant negative isoforms of p63 and for that matter of p73 are seen in vertebrates. These observations suggest an explosion of this gene family which must have gone along the lines of the following: the p63 gene assumes a new internal promoter that give rise to the ΔN-isoforms; this gene is duplicated to give rise to p73 which also has the TA and DN promoters and consequent isoforms. The homology relationships would demand that there was another gene duplication, this time of p73, to give rise to p53. Somewhere the p53 gene had to lose its internal promoter and several distal exons such that it was left with a single isoform that we know to be p53, the tumor suppressor. While possibly coincidence it is curious that p53 arose in evolution along with the ΔN-isoforms of p63 and p73, suggesting the very real possibility that some linkage was selected for based on their interaction. All of this forces a return to the epithelial stem cell as the potential site for the generation of the majority of cancers that afflict humans. Thus the stem cells for skin, breast, prostate, esophagus, airway epithelia, and urothelia all express large amounts of ΔN-p63 such that it is in large excess over that of p53. Whether that leads to a "p53-null" environment in the stem cell is unclear but might be at the basis of the unusual cell cycle potential of these cells. Our most recent estimates suggest that ΔN-p63 is present in 10-fold excess that of p53 and therefore could interfere or at least suppress any p53-like activity in these cells.
Conclusions and Perspectives
The advent of the p53 family has brought considerable excitement and speculation. The fundamental questions are whether p63 and p73 represent entirely novel pathways of tumor suppression or rather something more tied to p53 itself. Despite the strong evidence in favor of tumor suppressor actions of p73 and p63, the possibility remains that they are not tumor suppressors at all but in fact tumor promoters under certain pathological conditions. At present there is compelling evidence for both sides. Much of the mouse genetics done to date supports unique functions of these genes outside the parochial realm of cancer biology rather than their tumor suppressor actions. p63 appears essential for epithelial stem cell maintenance (32) while p73 governs a wide range of cellular responses including pheromone signaling, secretion, and inflammation. Most importantly, p73 null mice show no increase in rates of tumor formation suggesting no obvious link to tumor suppression (34). That being said, there is good evidence that p53 activity is defective in cells lacking p63 and p73 and that these genes play an integral role in the activity of our most important tumor suppressor. It is in view of this evidence that several groups have proposed to base gene therapy strategies on introducing p63 or p73 expression constructs to complement the mutation or loss of function of p53. Such approaches are in early stages but worth anticipating and developing. More concerning and possibly more physiologically relevant is the notion that ΔN-isoforms of p73 and p63 can impact negatively on p53 activity. The basis for this is already present in progenitor cell populations of many epithelial tissues where p63 is highly expressed. Whether this fact translates into early stages of tumorigenesis of these tissues is unknown at present, but the high expression of these potentially dominant negative factors clearly establishes a unique situation with regards to p53 activity. With increasing reports that various squamous cell carcinomas overexpress ΔN-p63, strategies to promote the degradation or transcriptional suppression of this gene, together with siRNA technologies, may herald new means of treating these recalcitrant tumors.
 XML Download
XML Download