INTRODUCTION
Arsenical compounds are distributed as natural toxicants, with no color, taste or smell. Arsenic trioxide, As2O3, has been reported to induce almost complete remission from acute promyelocytic leukemia (APL) (1). Cytopathological studies have also shown that As2O3 induces apoptosis in APL cells. Recent reports have shown that As2O3 down-regulates the bcl-2 gene expression and induces the expression of apoptosis related protein caspases, as well as degradation of PML and PML/PAR alpha proteins in APL cells. Similarly, arsenic trioxide suppresses the tumor cell growth by cell cycle arrest, cyclin-dependent kinase induction and apoptosis in MC/CAR myeloma cells (2). Arsenic trioxide-induced apoptosis is also enhanced in the presence of dithiothreitol in NB4 cells (3). Along with APL cells, arsenic trioxide can also induce apoptosis and cell growth inhibition in HPV16 infected cervical carcinoma cells (4). These collective studies suggest that arsenic trioxide has an anti-tumor activity in a variety of tumor cells. It has recently been reported that arsenic trioxide induces p53 accumulation via an ATM-dependent pathway in human fibroblast cells (5). Subsequently, p53 induces the expression of WAF1/p21 that has a growth-inhibitory ability, and elicits cell cycle arrests in the G1 phase, resulting in apoptosis (6). Furthermore, DNA breaks and DNA-protein cross-links are formed by arsenite treatments in a variety of cells (7). Although the exact mechanism(s) of arsenic trioxide to suppress cancer cell growth is still unclear, recent findings support the notion that it induces apoptosis of tumor cells, resulting in cancer cell growth inhibition. However, in one study, sodium arsenite (iAs3+) was found to promote cell growth and the expression of certain genes in bladder epithelial cells via activation of AP-1 transactivation (8). Clinically, arsenite should be tumor-specific and have low toxicity (9), so it is likely to be beneficial to develop more effective and less toxic arsenites for the treatment of cervical carcinomas. However, no studies on As4O6 have been reported.
Cervical carcinomas are mostly caused by infection with a high-risk group of human papillomavirus (HPV) (10). After a high risk HPV infection, two viral oncogenic proteins, E6 and E7, play a critical role in inducing cervical cancers by interacting with p53 and pRB for the inactivation of these cellular regulatory proteins, respectively (11). Current surgical, radiation, immune and gene therapies approaches have met with limited success. Furthermore, the early detection of cervical cancer using the Pap smear has contributed to a reduction in the incidence of cervical cancers. However, relapsing cervical cancers have been problematic, adding importance to the development of anti-cervical cancer drugs.
The recent development of cDNA microarray technology has allowed to the simultaneous monitoring of the expression profiles of thousands of genes. Also, the annotation project directed by the Gene Ontology (GO) Consortium (http://www.geneontology.org) has been used to quantitatively understand multiple relationships between differentially regulated genes and pathogenesis. Gene expression profiles obtained from patients from cDNA microarray analyses could be useful for predicting the outcome of chemotherapy as well as the duration of patient survival. A generated fingerprint of gene expression profiles could be useful in elucidating cellular changes for any anti-cancer drug treatment. This is likely to shed light on the determining effects of chemotherapeutic regimens on tumor suppression.
Herein, a new arsenical compound, As4O6, has been designed and its ability to suppress cell growth and induce gene expression patterns tested using a cDNA microarray in HPV16 immortalized cervical carcinoma cells, SiHa cells, and compared to As2O3. It was observe that As4O6 was more effective in suppressing the SiHa cell growth at a lower dose compared to that of As2O3. Differential gene expression profiles of SiHa cells were also observed on treatments with As2O3 and As4O6, suggesting that further investigation of the identified genes is warranted. Thus, these data suggest that As4O6 could be a more potential drug candidate for the treatment of cervical cancers.
MATERIALS AND METHODS
1) Cell culture
Immortalized HPV16 human cervical carcinoma cell lines, SiHa cells containing wild type p53, were incubated in DMEM, supplemented with 5% fetal bovine serum, 0.37% sodium bicarbonate, 30 mM HEPES and 100 µg/ml streptomycin/penicillin (cDMEM), at 37℃ in a CO2 incubator.
2) Chemical reagents
The As2O3 was purchased from Sigma and the As4O6 was synthesized and provided by Chonjisan., Co., Seoul, Korea. These chemicals were diluted in H2O to a final concentration of 10-3 M and kept at 4℃. MTT was purchased from Sigma and dissolved in PBS to a final concentration of 5 mg/ml.
3) DNA fragmentation assay
SiHa cells were divided into 5×105 cells per 100 mm dish plate. After 24 hr incubation, different amounts of arsenic compounds were added to the cells, which were incubated for a further 48 hrs at 37℃. The cells were then centrifuged and lysis buffer [0.8% SDS, 0.1 M NaCl, 0.1 M EDTA, 50 mM Tris-HCl (pH8.0)] added to the resultant cell pellets, followed by the addition of 20 µg/ml proteinase K (Sigma). This mixture was incubated for 4 hrs at 56℃. DNA was extracted by phenol/chloroform treatment. Five µg of extracted DNA was analyzed on a 2% agarose gel containing ethidium bromide (0.1 µg/ml) and the DNA ladder formation visualized under UV light.
4) Cell growth inhibition assay
As a cell growth inhibition assay, the MTT assay was performed. First, SiHa cells (1×103 cells/well) were divided into a 96 well plate in 100 µl of cDMEM. After 24 hr incubation, the cells were treated with a different amounts of arsenic compounds. After 4 day incubation, 20 µl of MTT solution (Sigma, St. Louis, MO) was added to each well of the plates, which were then incubated for 4 hrs at 37℃. The cell media were replaced with 100 µl of DMSO (Sigma) per well and the plates shaken for 10 sec. The optical density (OD) was then measured at 570 nm using an ELISA-Reader (Spectromax 250, Molecular Devices). The growth inhibition rate (%) was calculated as follows: OD of non-treatment -OD of drug treatment/OD of non-treatment×100.
5) RNA and probe DNA preparation
Total RNA was prepared using Trizol reagent (MRC, Cincinnati, Ohio), according the manufacturer's protocol. For the RT reaction, 50 µg of total RNA was mixed with 1 µl of control mRNA (lambda bacteriophage mRNA, 0.5 µg/ul) and 1.5 µg of oligo dT primer (1.5 µg), to a final volume of 20.5 µl. The reaction mixture was heated for 5 min at 70℃, and the denatured RNA reacted for 1 hr at 42℃ in AMV buffer (low dT NTP, Cy3 or Cy5-dUTP, RNase inhibitor and AMV reverse transcriptase). Ten µl of 1 M NaOH was added to the reaction mixture for 10 min at 37℃, followed by the addition of 25 µl of 1 M Tris- HCl (pH 7.5). Probes were purified using Sephacryl S-100 columns And the ethanol-precipitated DNA probes solubilized in 15 µl of hybridization buffer (6×SSC, 0.2% SDS, 5×Denhardt solution and 0.1 µg of salmon sperm DNA).
6) Probe hybridization
The probes were denatured for 2 min at 95℃ and then kept on ice. These were centrifuged for 10 min at 14,000 rpm, and the supernatant containing probes added drop wise to a 384 cDNA chip (Macrogen, Seoul, Korea). After sealing with a cover glass, the hybridization reaction was performed for 12~16 hrs at 62℃. After hybridization, the slides were washed twice for 30 min in 60℃ washing buffer (2×SSC, 0.2% SDS) and then dried at the room temperature.
7) Northern blotting
Twenty micrograms of each RNA sample were electrophoresed through 1.1% formaldehyde-agarose gels and transferred to Hybond-N nylon membranes (Amersham Pharmacia Biotech). First strand cDNAs were incorporated with 32P-dATP during reverse transcription from total RNAs using oligo (dT) as primer. Hybridization was carried out overnight at 68℃ in Rapid Hyb-buffer (Amersham Co.). After hybridization, the membrane was washed twice at room temperature in 2×SSC and 0.1% SDS, and a further twice at 65℃ in 0.1×SSC and 0.1% SDS and then radiophotographed at -70℃ for 24~48 hr.
8) Scanning and data analyses
The DNA chip was scanned using a Generation III Array Scanner (Amersham Pharmacia Biotech.) and then analyzed via ImaGeneTM Version 3.0 (TaKaRa, Kyoto, Japan), according to the manufacturer's protocols. To classify the gene profiles into the gene ontology, cellular process analysis was carried out, as previously described (12). All files, including the results of the microarray experiment and the Gene Ontology analysis were downloaded from our anonymous FTP site: ftp://160.1.9.42/work/arsenic/
RESULTS
1) Both As2O3 and As4O6 induced apoptosis in SiHa cell lines
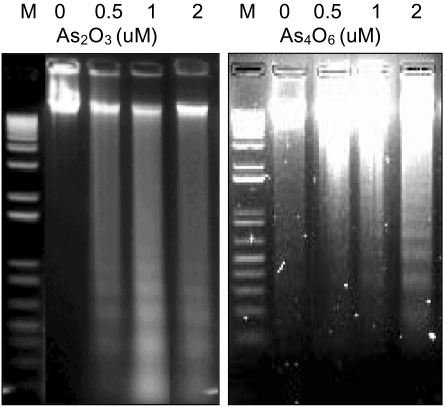
To investigate whether a new arsenical compound, As4O6, could induce apoptosis in SiHa cells, increasing amounts (0.5, 1 and 2 µM) of As4O6 were used to treat the cells for 48 hrs. As a positive control, cells were also incubated with As2O3. As shown in Fig. 1, both As2O3 and As4O6 induced DNA ladder formation at all concentrations tested (0.5, 1 and 2 µM), suggesting that As4O6 can also induce apoptosis in SiHa cells.
2) Comparison of effects of As2O3 and As4O6 on SiHa cell growth in vitro
To the compare anti-growth effects of As2O3 and As4O6 in SiHa cells, increasing amounts of arsenical compounds were added. As shown in Fig. 2, the growth of SiHa cells was decreased by the increasing amounts (0.5, 1, 2, 3, 4 and 5 µM) of both As2O3 and As4O6, as determined by the OD values. In the case of As2O3, the 1 µM treatment resulted in approximately 50% cell growth inhibition over the time periods, whereas the doses from 2 to 5 µM showed complete suppression of cell growth. In the case of As4O6, however, the 0.5 µM treatment resulted in approximately 50% cell growth inhibition over the time periods, whereas the doses ranging from 1 to 5 µM showed complete suppression of the SiHa cell growth. The cell growth inhibition rates (%) of As2O3 were calculated as 9.2, 56, 89, 93, 96 and 96% at concentrations of 0.5, 1, 2, 3, 4 and 5 µM, respectively. In contrast, the cell growth inhibition rates (%) of As4O6 were calculated as 54, 84, 84, 85, 85 and 87 at concentrations of 0.5, 1, 2, 3, 4 and 5 µM, respectively.
3) Differentially regulated gene expression profiles of SiHa cells by As2O3 and As4O6
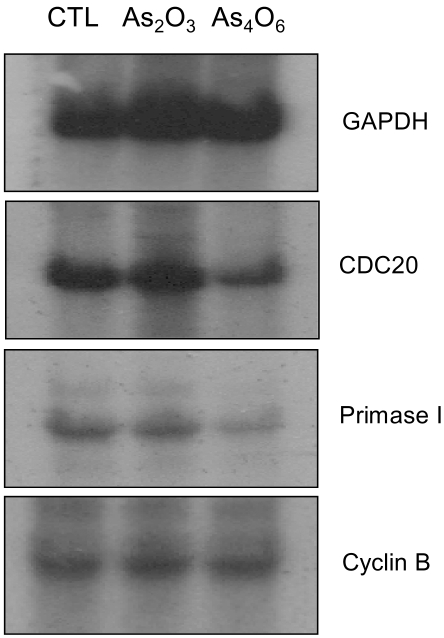
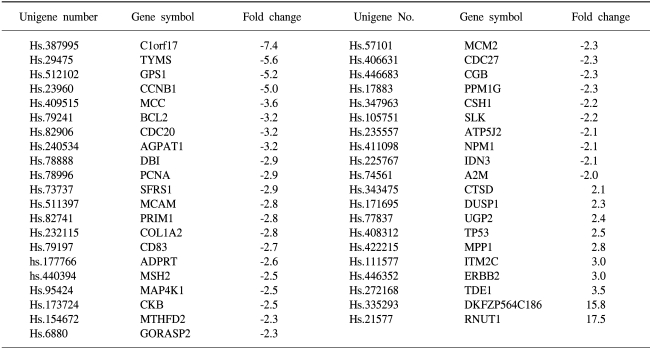
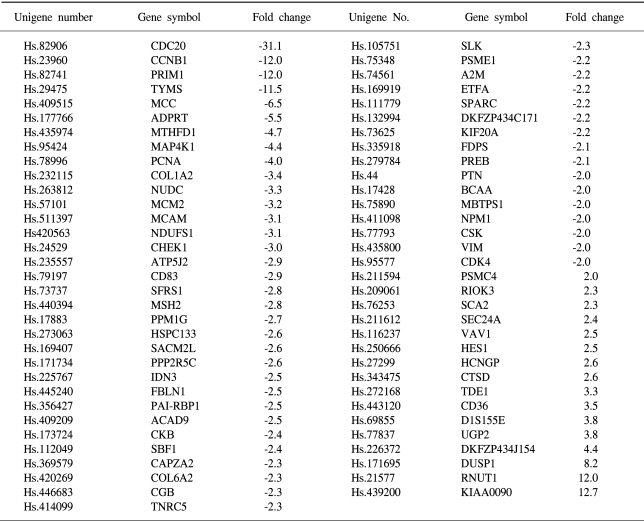
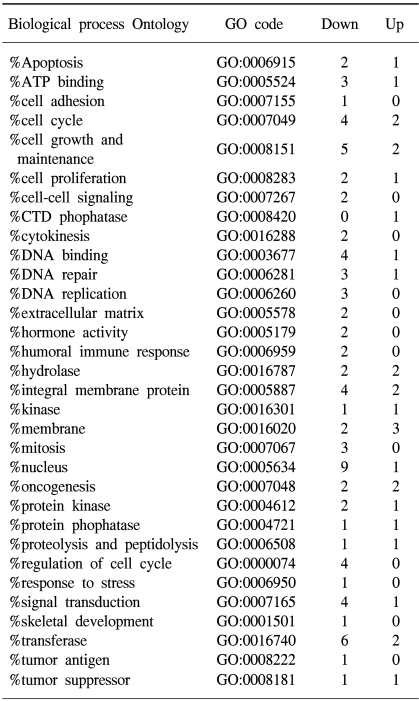
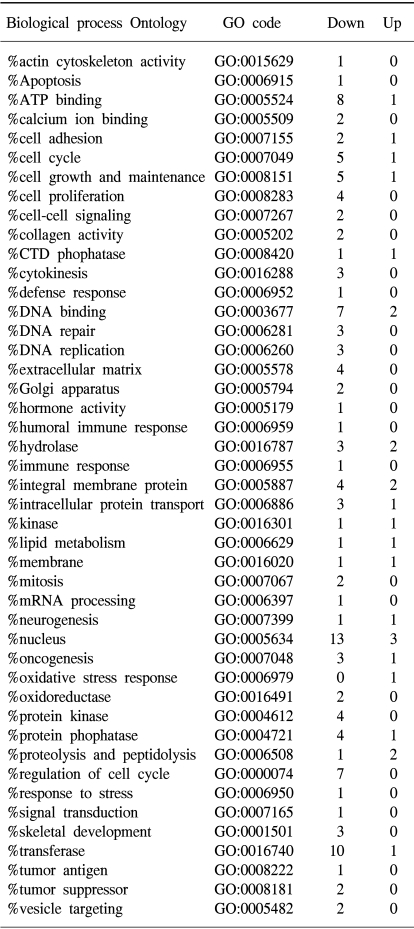
To determine the gene expression patterns of SiHa cells due to As2O3 and As4O6 treatment, the cells were incubated with 1 µM of As2O3 or As4O6, and their gene expression profiles analyzed using the 384 cDNA microarray system and compared to cells with no drug treatment. Northern blots were performed to confirm the gene expression patterns, and showed the consistency of the assays (Fig. 3). Table 1, 2 show the differentially expressed gene expression profiles due to the As2O3 and As4O6 treatments, respectively. More gene expressions were up- or down-regulated by As4O6 compared to As2O3. With the As2O3 treatment, 41 genes were more than 2 fold up- and down-regulated in their expressions. In the case of As4O6, 65 genes showed differential expressions greater than 2 fold. Among these genes, 5 were enhanced in their common expressions by both As2O3 and As4O6 treatments. These genes might possibly be associated with resistance to the arsenical drugs. These included UDP-glucose dehydrogenase, tumor differentially expressed 1, dual specificity phosphatase 1, cathepsin D and snuportin-1. In particular, 22 genes were commonly down-regulated by both As2O3 and As4O6. This suggests that these identified genes might be associated with drug sensitivity in SiHa cells. These included CDC20 (cell division cycle 20), cyclin B1, thymidylate synthetase and primase, as well as others (Table 3). In particular, the expressions of genes coding for thymidylate synthetase, primase, cyclin B1 and CDC20 were highly down-regulated by the As4O6 treatment.
The Gene ontology classification of the differentially expressed genes revealed that most were functionally related to apoptotic regulation, as shown in Tables 3 and 4. The cellular processes identified as significant by the GO are listed in the Tables, including the expression patterns. There were several significant differences between the expression profiles of the functions in As2O3 and As4O6, including 3 significantly up-regulated genes coding for the proteins involved in membrane activity, 1 coding for oxidative stress response, 2 for proteolysis and peptidolysis, and other miscellaneous genes. In contrast, significantly down-regulated genes were found coding for DNA metabolism, humoral immune response, nucleus activity, cell cycle regulation and cell proliferation, and other miscellaneous genes. In particular, genes coding for the proteins involved in cell cycle and nucleic acid metabolism, such as CDC20 (cell division cycle 20) and cycline B1, had completely decreased expressions.
DISCUSSION
The anti-tumor functions of the arsenic compound, As2O3, have been reported in leukemia both in vivo and in vitro (1). It was also observed that As2O3 induced apoptosis and cell growth inhibition in SiHa cell lines. This supports the previous findings that arsenic trioxide induces anti-tumor effects through the induction of tumor cell apoptosis (2). In the case of promyeloleuckemic cells, As2O3 down-regulates the expressions of bcl-2 and PML/RARα/PML protein, and are correlated with apoptosis (1). As2O3 also induces the production of p53 protein (5). The p53 protein subsequently induces cell cycle arrests and apoptosis through the induction of the WAF1/p21 expression (6). It was also reported that the glutathione redox system is associated with apoptosis due to As2O3 (13). It is likely that apoptosis is a mechanism by which arsenic trioxide suppresses tumor cell growth both in vivo and in vitro. This is also supported in that As2O3 was also found to inhibit cell growth and induce apoptosis in different cancers, including leukemia, myelomas, lymphomas and solid tumors (14).
In contrast to As2O3, As4O6 was synthesized and evaluated for its anti-tumor effects in SiHa cells. As with As2O3, As4O6 also induced apoptosis and cell growth inhibition in vitro. In particular, As4O6 showed a significant anti-tumor activity (approximately 50% growth inhibition) at a dose of 0.5 µM, at which As2O3 displayed little anti-tumor growth effects. Instead, 1 µM of As2O3 showed approximately 50% growth inhibition, suggesting that As4O6 could be more potent in the control of cervical cancer cell growth. However, with doses ranging from 2 to 5 µM both As2O3 and As4O6 displayed a complete anti-growth effect. The dose effects of arsenic trioxide on growth inhibition are consistent with many previous reports (2,4). As4O6 was also injected into mice, along with As2O3, in order to compare their toxicities in animals. Mice injected with As4O6 survived longer than those injected with As2O3 (data not shown), suggesting that As4O6 is less toxic to animals than As2O3. These support the notion that As4O6 could be a less toxic, but more potent drug for controlling cervical cancers.
The cDNA microarray was further used to compare the gene expression profiles of these two drugs in SiHa cells. A new classification of cancer cell types has been proposed based on the altered gene expressions of cancer cells (15). It has also been reported that gene expression profiles might reflect drug resistance or sensitivity in tumor cells (16). Recently, cDNA microarray analyses showed a dramatic difference in the gene expressions between normal liver tissues and liver tissues from arsenic-exposed patients (17). The differentially expressed genes included those involved in cell-cycle regulation, apoptosis, DNA damage response and intermediate filaments. A significant difference in the gene expression profiles was also observed between As2O3 and As4O6. For example, As2O3 influenced the expression of a total of 41 genes, whereas As4O6 influenced a total of 65 genes. This correlates well with the observed difference in the cell growth inhibitory abilities of these two drugs. These data also support the notion that As4O6 behaves differently from As2O3. Interestingly, the activity of response to oxidative stress (DUSP1) involved in the metabolism was completely up-regulated by As4O6. However, commonly regulated genes, such as up-regulated genes (UDP-glucose dehydrogenase, tumor differentially expressed 1, dual specificity phosphatase 1, cathepsin D and snuportin-1) and down-regulated genes (CDC20, cyclin B1, primase polypeptide 1, thymidylate synthetase, proliferating cell nuclear antigen) were also observed in the presence of both As2O3 and As4O6.
The functions of up-regulated genes are unclear, but the Gene Ontology analysis of the differentially regulated genes revealed that most of down-regulated genes play key functions in various cellular activities involved in cell death. Genes associated with DNA and cellular metabolisms are commonly down-regulated compared to their counterparts. The regulation of extracellular matrix expression is a key to the tissue remodeling in normal and pathologic processes that lead to tissue carcinogenesis (18). Interestingly, the genes in this cellular process were highly down-regulated by both arsenic compounds. Among the genes in this category were the collagens. The expression of collagens I that regulates its turnover has been demonstrated in tumorigenesis (19). Cytoskeleton integrity plays an important role in the cell cycle progression, cell death and cell differentiation. An abnormal cytoskeleton is often observed in cancer cells. In this study, the gene expression profiles involved with the cytoskeleton were completely down-regulated. Disruption of cell cycle progression pathways has been implicated in abnormal cell growth and carcinogenesis (20). In this study, it was observed that genes involved in the cell cycle, such as CCNB1 and MCM2, were highly down- regulated.
Cyclins, known as cell cycle regulators, bind to and activate the CDKs that are also regulators of the cell cycle in eukaryotic cells. The activation and subsequent inactivation of cyclins and CDKs are important in the control of the cell cycle (21). CDC20 is a regulator in mitotic (anaphase) checkpoints (22). Cyclin B1, a cell cycle checkpoint protein, regulates the transition from the G2 to M phase (23). Cyclin B1 protein has recently been reported to be a shared epithelial tumor associated antigen, showing a high error rate in the translation of cyclin B1 proteins in tumors. Similarly, primase has an important function in DNA replication, DNA repair and cell cycle checkpoints (24). Thymidylate synthetase catalyzes the conversion of dUMP to thymidylate, which is an essential component of DNA. Inhibition of this enzyme activity results in cell death. Proliferating cell nuclear antigen is also associated with DNA repair processes. In particular, the level of proliferating cell nuclear antigen correlates with DNA repair activity (25). In our cDNA microarray analyses, these cell regulatory genes (CDC20, cyclin B1, primase, thymidylate synthetase and proliferating cell nuclear antigen) were down-regulated by both As2O3 and As4O6. It is likely that these genes are associated with arsenite-mediated apoptosis and growth inhibition in SiHa cells. Down-regulation of the genes involved in the cell cycle, DNA repair and regulation was previously reported in arsenic-exposed patients. This further supports that these arsenical compounds might inhibit tumor cell growth at least through inhibition of expression of these regulatory genes. It also appears that As2O3 and As4O6 induce anti-tumor effects through a unique, but overlapping regulatory mechanism(s). This is based on our findings that two arsenical drug compounds displayed differential gene expression patterns with some commonly expressed genes.
CONCLUSIONS
The data presented herein suggest that As4O6 is more effective than As2O3 for inhibiting the growth of HPV16 infected SiHa cervical cancer cells. These two drugs appear to mediate the tumor cell growth inhibition at least through a unique, but overlapping regulatory mechanism(s), as determined by cDNA microarray and Gene Ontology analyses, reflecting evidence that indicates similarities and differences between the molecular environments of the cell growth suppression pathways. Thus, this study is likely to provide a new potential drug approach for treating cancers in clinical settings.