

 PDF
PDF Citation
Citation Print
Print


Fibrohistiocytic tumours are well-recognized histopathological entities that usually occur in subcutaneous and soft tissues. Nevertheless, such lesions can also involve the neural axis. We recently encountered a particularly curious example arising from the pachymeninx; thus, we present this unique case along with its singular morphological traits.
CASE REPORT
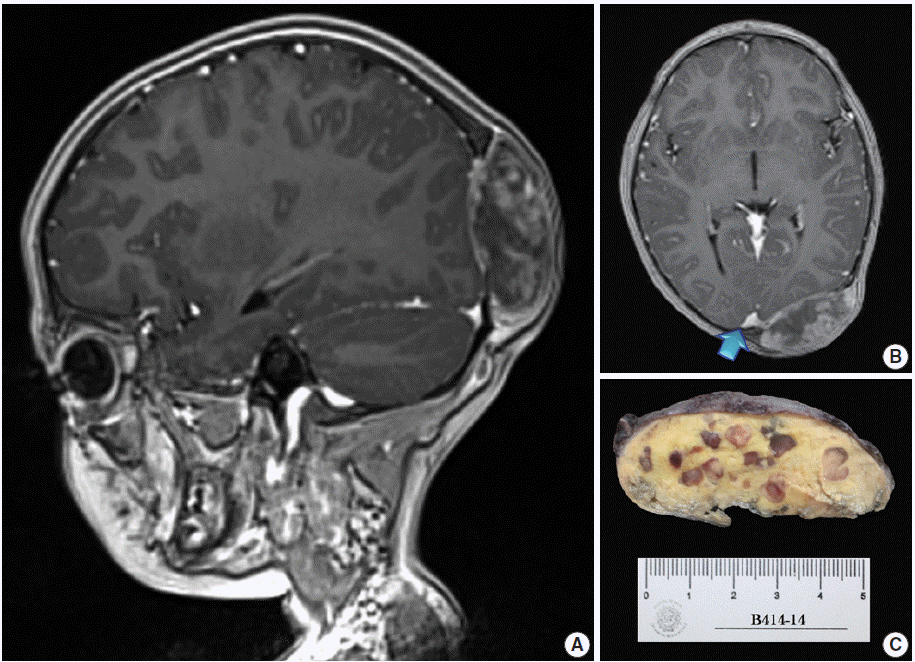
A 4-year, 7-month-old girl presented for medical consultation with her parents. They complained about a lump in the back of the child’s head that made it difficult to comb and plait the girl’s hair. The child was entirely asymptomatic, and there was no relevant medical history, such as systemic ailment or associated long-bone disease. Haematological and biochemical tests were unremarkable, and no lipid abnormality was detected. Magnetic resonance imaging scans showed an occipital dural-based tumour of heterogeneous intensity slightly compressing the left occipital pole and apparently extending toward the superior sagittal sinus (Fig. 1A, B). Resection was performed, and the surgical specimen was submitted for histopathological evaluation.
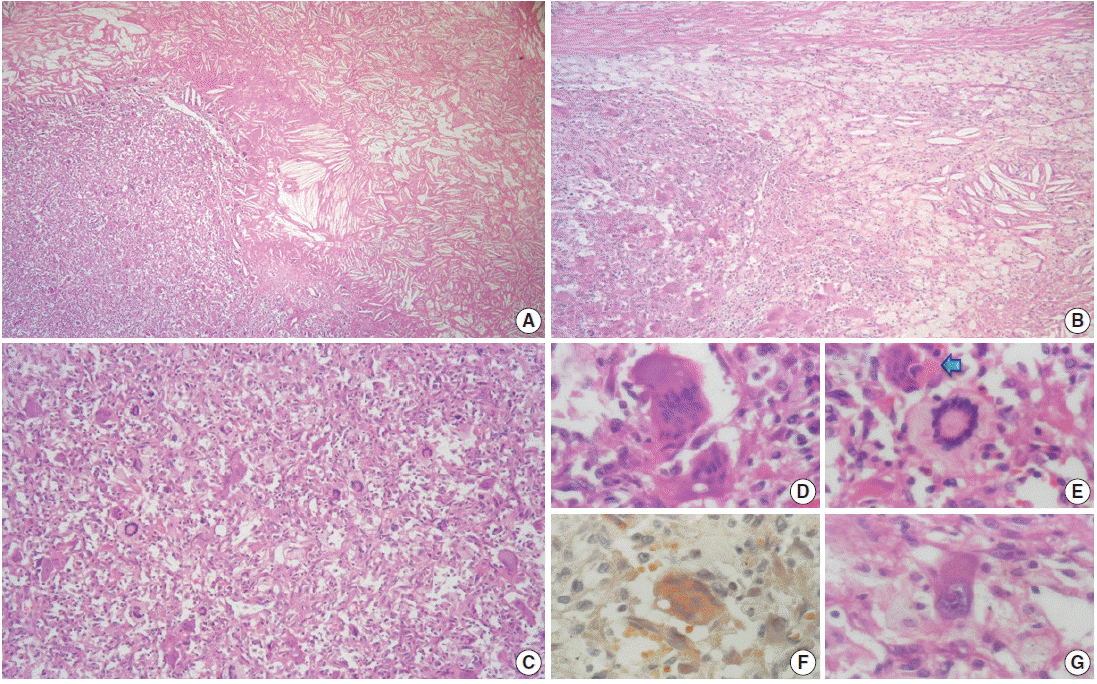
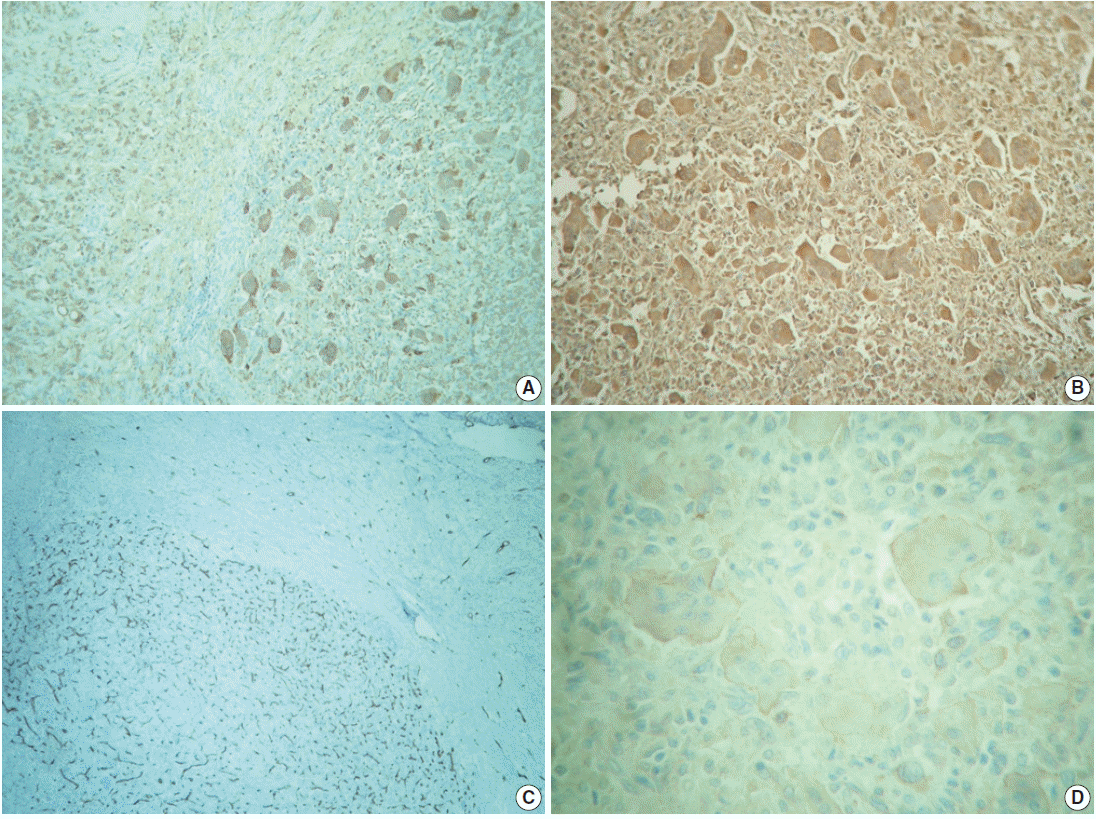
The outer surface of the fixed specimen (6×6×2 cm) was gray tan, smooth, and hard, yet the transverse cut surface contained yellow, pale, and pasty material with reddish kernel-like crusts, resulting in an overall appearance similar to a slice of butter-covered raspberry bread (Fig. 1C). Under light microscopy, the pale yellow areas contained widespread coagulative necrosis, powdery calcification, numerous cholesterol clefts, foreign body-type giant cells, and foamy macrophages, while the red nodules were populated by giant cells (Fig. 2A–C). The entire lesion was demarcated by a thin fibrous wall consistent with compacted dura mater. Closer examination of the nodular areas detailed a miscellaneous group of giant cells including mononucleated as well as multinucleated forms of foreign-body and Touton types, the former with ground-glass amphophilic to flamboyantly eosinophilic cytoplasm and occasional empty vacuoles (Fig. 2C–G). At times, foci of a mixed inflammatory cell population could be recognized, including lymphocytes, neutrophils, and scarce eosinophils. Intriguingly, some giant cells demonstrated phagocytosis of related tumoural elements (cell cannibalism) and uptake of inflammatory cells (Fig. 2E). Disturbing bizarre nuclei were also present in some of these giant cells; however, mitotic activity was not identified (Fig. 2G). The immunoperoxidase-coupled reactions revealed a fibrohistiocytic immunophenotype with CD68, α1-antitrypsin (data not shown), α1-antichymotrypsin (data not shown), and vimentin positivity (Fig. 3A, B). CD34 immunostaining was negative but highlighted the delicate vascular branching, whereas the epithelial membrane antigen (EMA) unexpectedly delineated the giant cell borders (Fig. 3C, D). Likewise, staining for PS100, CD1a, CD207, FXIIIa, smooth-muscle actin (SMA), muscle-specific actin (MSA), podoplanin (D2-40), microphthalmia transcription factor (MITF), and glial fibrillary acidic protein (GFAP) was also negative. Ancillary histochemical stains were also requested (Ziehl-Neelsen, auramine-rhodamine, Fite-Faraco, periodic acid-Schiff, Brown-Hopps and Grocott) and did not detect either conventional or atypical mycobacteria. An additional dural fragment labeled as “implant attached to the superior sagittal sinus” was received. It had yellow macules constituted microscopically of foamy histiocytes and multinucleated foreign-body type giant cells engulfing cholesterol crystals.
According to the aforementioned findings, the lesion was diagnosed as dural xanthogranuloma (cholesterol granuloma with broad areas of coagulative necrosis and foci of giant cells). To date the patient is healthy and without any complaints.
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from the patient’s legal guardian, and anonymity of the patient was preserved.
DISCUSSION
Lesions of fibrohistiocytic origin seem to be quite rare among central nervous system (CNS) tumours [1-3]. Accumulation of foamy macrophages in the neural axis can arise from known metabolic/storage disorders or serum lipid disturbances (secondary CNS involvement) but can also occur without an exact cause or triggering factor (primary CNS involvement) [4-7]. At present, the pathogenesis of the latter remains poorly understood. Nonetheless, two hypothetical pathways have been proposed to explain their formation [5-7]: (1) local trauma or haemorrhage with overstimulation of mesenchymal cells from dura mater or vascular adventitia and (2) synthesis of lipotrophic factors that cause undifferentiated mesenchymal cells to undergo xanthomatous transformation. These primary CNS, lipidized fibrohistiocytic tumours have been traditionally dichotomized as intraventricular (i.e., affecting plexi choroidei) or meningeal (dural-associated) [4-7]. This classification can be further expanded to include some other topographies, such as leptomeningeal or parenchymatous (intracerebral or intracerebellar) [8-10].
Although plexi choroideorum xanthomas have been described since the beginning of the 20th century [11], the first observations of a non-ventricular fibrous xanthoma were presented in the 1973 work of Kepes et al. [10]. It was not until 1979 that Lam and Colah [12] reported the first dural-associated lesion of this kind. In contrast, CNS cholesterol granulomas (xanthogranulomas) are uncommon lesions usually found incidentally in plexi choroidei during postmortem examination (1.6% to 7% of autopsies). They are thought to occur either because of desquamation of choroidal epithelial cells or from local histiocytes recruited, perhaps, as formerly described [13]. They feature needle-like cholesterol clefts flanked by foreign-body type giant cells, foamy histiocytes, chronic inflammatory cells, and scattered haemosiderin granules [1,2]. Also, varying amounts of fibrosis can be present, becoming most marked in long-standing lesions. Otherwise, solitary reticulohistiocytoma is a distinctive but rare lesion in adults [1,14,15]. Such tumours are seen as a yellow-brown to dark-red papule consisting of dense circumscribed nodules of deeply eosinophilic histiocytes often exhibiting multinucleation with no Touton giant cells. Some degree of nuclear atypia and occasional mitotic figures might be present. Immunoperoxidase reactions reveal expression of CD163, CD68, and vimentin, along with variable reactivity for HAM56, α1-antitrypsin, lysozyme, FXIIIa, PS100, MSA, and MITF. A multicentric variant has also been described, but it is associated with autoimmune disorders and internal malignancies. Interestingly, it has been suggested that the solitary form is similar to adult xanthogranuloma, with the distinction being largely based on predominance of multinucleated eosinophilic histiocytes [1]. Reports of solitary reticulohistiocytoma involving the neuroaxis are currently non-existent in the global medical literature.
There were various differential diagnoses to consider in our case, such as xanthoma, fibrous histiocytoma (fibroxanthoma), atypical fibrous histiocytoma, solitary (juvenile) xanthogranuloma, and Erdheim-Chester disease. As conveyed by Weiss and Enzinger, xanthomas are just collections of tissue histiocytes filled with lipids [1,5,7]. These foamy macrophages are embedded within a loose stroma and organise in uniform sheets, occasionally divided into smaller nests by delicate fibrous bands. They are thought to be closely related to cholesterol granulomas, probably in an earlier stage. Nevertheless, a comparative clinicopathological analysis of both lesions in plexi choroidei revealed some differences [16]. Intense siderosis and a higher male-to-female ratio were noted in cholesterol granulomas (14:3 vs 13:8), while xanthomas were more frequently associated with dyslipidaemia. Histologically, benign fibrous histiocytomas are characterized by a mixture of spindle fibroblast-like cells arranged in short fascicles, with or without focal storiform profiles, and plump histiocyte-like cells accompanied by variable numbers of foamy histiocytes, haemosiderin-laden macrophages, multinucleated giant cells of foreign-body or Touton types, lymphocytes, plasma cells, and bundles of collagen [1-3]. The immunohistochemical profile shows an admixture of CD68, FXIIIa, CD34, PS100, SMA, and D2-40 cells, in variable proportions [1,3]. A subset of fibrous histiocytomas exhibits borderline histologic signs of malignancy, including significantly greater cellular atypia and increased mitotic activity [1]. In spite of the nuclear pleomorphism focally detected in our case, mitotic activity was not present; thus, the bizarre nuclei observed were considered a degenerative phenomenon such as that seen in ancient neurilemmoma or giant cell ependymoma. We also interpreted the small implant attached to the superior sagittal sinus as an additional lesion rather than direct extension from the larger one (i.e., an early multifocal process). Juvenile (solitary) xanthogranuloma demonstrates a wavering time-dependent appearance and is a challenging diagnosis to eliminate [1-3,17]. Despite its classic morphology showing foamy histiocytes accompanied by Touton-type giant cells, early lesions tend to possess less intracytoplasmic lipid droplets. Hence, mononuclear cells display a plump eosinophilic cytoplasm. Conversely, longstanding lesions usually develop interstitial fibrosis and even assume a vague storiform pattern. Additionally, the existence of transitional cases with or without xanthomatous features, with or without interspersed spindle cells, and containing multinucleated giant cells with or without Touton configuration is also possible. Usually, a modest number of acute and chronic inflammatory cells are also present, especially eosinophils. Regardless of morphology, macrophages and Touton cells show immunoreactivity for CD68, α1-antitrypsin, α1-antichymotrypsin, lysozyme, CD31, and FXIIIa. Langerhans and non-Langerhans cell histiocytoses are also worth considering [2]. The former can be ruled out by means of an appropriate immunohistochemistry panel (PS100, CD1a, and CD207) in a suitable clinical setting (localized vs multiple sites of involvement). On the other hand, the latter can be troublesome, particularly when Erdheim-Chester disease is suspected, which is a rare disorder that most often becomes apparent in middle age and is typically seen in the context of long-bone and systemic disease. It is characterized by lipid-laden CD68+, CD1a–, and PS100– macrophages and Touton-type multinucleated giant cells accompanied by scarce eosinophils. Indeed, the reddish nodules described in our case displayed some of these features; however, considering the lack of multiple organ involvement, we considered the multinucleated foreign-body type giant cells to represent a trait more akin to solitary reticulohistiocytoma than to Erdheim-Chester disease. Tuberculosis was also a consideration; however, the gross appearance of the lesion was not consistent with a tuberculoma, which is usually hard and chalky-white rather than soft, and pale-yellow. Also, tuberculosis tends to be infratentorial in the paediatric population. Furthermore, there was no Heubner arteritis or typical epithelioid granulomas of tuberculosis with caseous necrosis and Langhan type giant cells. One might argue that it can be entirely feasible in an immunodeficient patient; however, as previously stated, our patient was a completely healthy girl who had already received her bacillus Calmette–Guérin vaccine (which only provides protection against the leptomeningeal form). Other non-fibrohistiocytic neoplasms can show extensive xanthomatous changes, such as glioblastoma, pleomorphic xanthoastrocytoma, and metaplastic meningioma [10,18]. Nonetheless, immunoperoxidase-coupled assays for GFAP and EMA can be useful to achieve a diagnosis in these settings. Though a dull peripheral rim was observed in the multinucleated giant cells through EMA immunostaining, it was uncertain whether to dismiss it as an artifact. Indeed, EMA expression has been demonstrated in normal plasma cells, monoblasts, and in some hematological malignancies [19].
Xanthogranuloma and solitary reticulohistiocytoma are conditions grouped among the pathogenetically diverse category of fibrohistiocytic tumours [1]. Interestingly, these miscellaneous lesions sometimes show stunning overlapping morphological or immunophenotypical traits, which might be explained on the basis of the “facultative fibroblast” concept proposed by Ozzello in 1963 [12,20]. This concept features a cell with an inherent capacity to transdifferentiate to another morphologic and functional state, an assumption that could be true for the present case. Nonetheless, it can simply represent a time-dependent phenomenon (i.e., evolving stages of the same process). Hence, we report a lipidized fibrohistiocytic lesion arising in the dural coat of the CNS with peculiar and unconventional traits that, as far as we know, have not been previously described.
 XML Download
XML Download