

 PDF
PDF Citation
Citation Print
Print


Analogous to the mixed epithelial/mesenchymal tumours seen in Müllerian derivatives, some glial neoplasms can display a bimorphic appearance due to an accompanying mesodermal component. While gliosarcoma is probably the best-known paragon of this group, the existence of non-malignant mesenchymal constituents, different from the so-called Scherer’s tertiary structures, is rare but feasible. Here, we describe a peculiar case of a high-grade astrocytic neoplasm (glioblastoma) blending with an innocuous spindle-cell component.
CASE REPORT
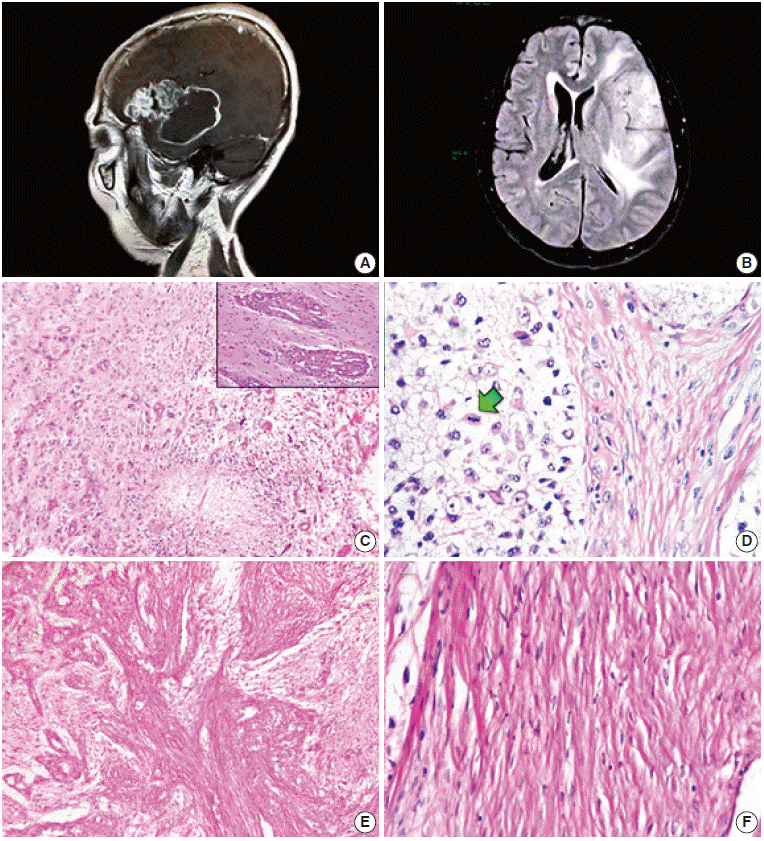
A 50-year-old female presented with headache and loss of memory over the previous six months. Magnetic resonance imaging scans showed a left parieto-temporal mass (70.82×48.56 ×43.97 mm) with significant perilesional oedema and post-contrast annular enhancement (Fig. 1A, B). Despite prompt tumour sampling and analysis, the patient ultimately expired one month after the surgery. Necropsy was not authorized.
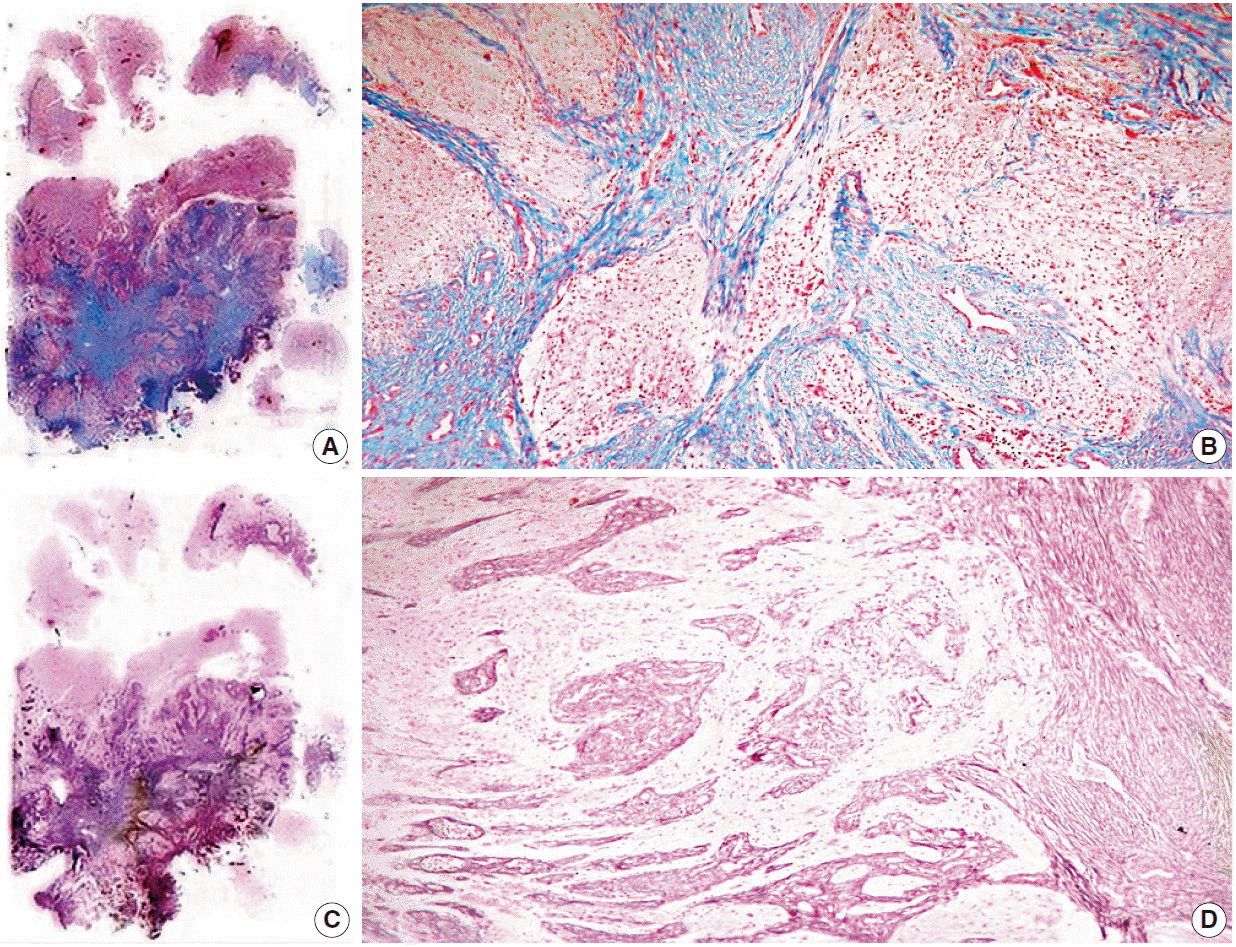
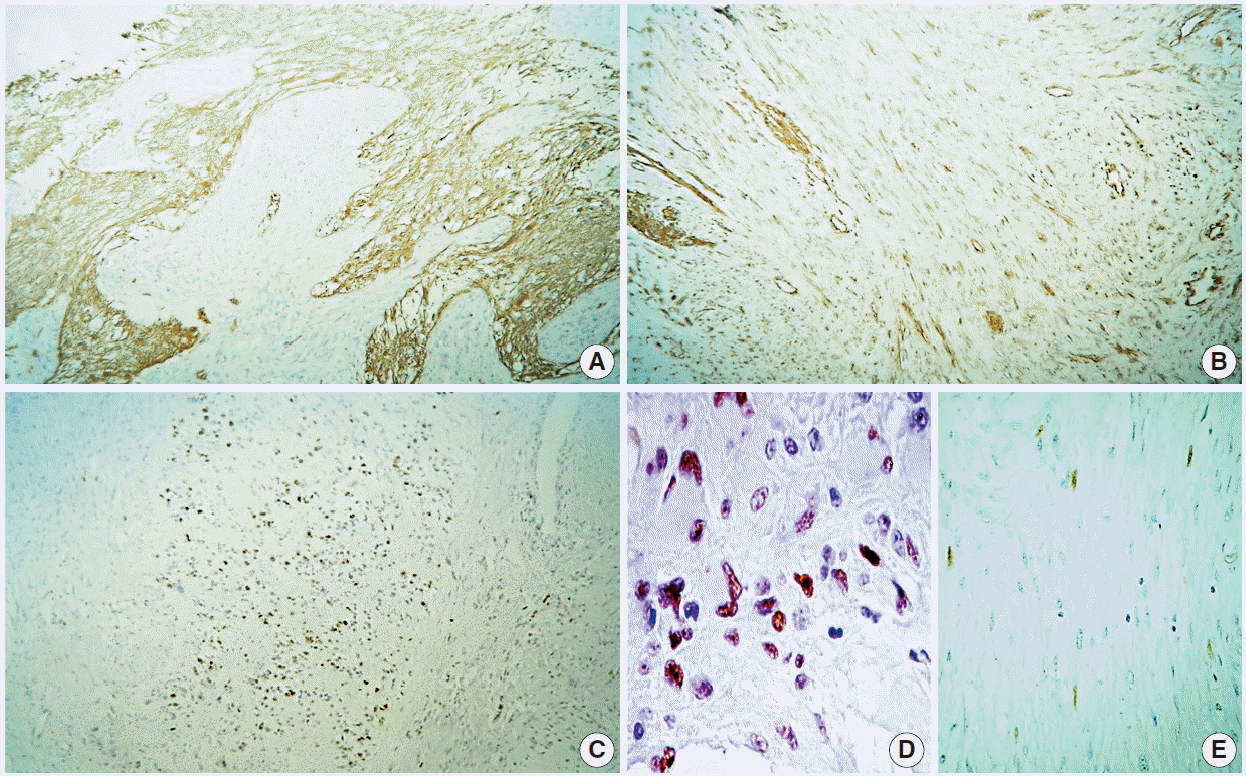
Histologically, the lesion showed areas unequivocally consistent with glioblastoma due to their microvascular proliferation, palisading necrosis, hypercellularity, pleomorphism, and high mitotic activity (Fig. 1C, D). These areas intermingled with cytologically bland, long spindle-cell fascicles (Fig. 1E, F) enveloped by sturdy collagen and fine reticulin fibers (Fig. 2A–D). As expected, immunoperoxidase-coupled reactions showed strong and diffuse expression of glial fibrillary acidic protein in the glioblastoma areas (Fig. 3A), while the spindled part of the neoplasm was exclusively positive for vimentin (Fig. 3B). Ki-67 expression was variable, with positivity as high as 50% in the astroglial regions (Fig. 3C) but with immunolabeling of less than 1% in the mesenchymal portion. Likewise, expression of p53 was largely diffuse in the gliomatous parts, albeit focally in some fusiform cells (Fig. 3D, E). No reaction was detected for epidermal growth factor receptor in either element.
The lesion was diagnosed as gliofibroma (mixed astrocytic neoplasm with a low-grade mesenchymal constituent), with special note of its poor prognosis based on the degree of anaplasia of the glial component, suggestive of glioblastoma. This note was written prior to the patient´s demise.
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from the patient’s legal guardian, and the anonymity of the patient was preserved.
DISCUSSION
The term gliofibroma was first introduced by Friede in 1978 [1] to describe an unusual neoplasm featuring astroglial cells within collagen bundles. Since then, nearly 40 cases of gliofibroma have been reported in the worldwide literature, the vast majority occurring in the paediatric population [2]. Only 11 cases have been recorded in adults, with age ranging from 19 to 54 years [3,4]. More importantly, some cases described in both, children and adults, bear morphological traits indicative of biologically aggressive lesions (Table 1). The prognostic value of these features in gliofibromas has been widely debated, as some non-lethal tumours have a high-grade glial component while other are recurrent lesions despite their low-grade glial associate [5,6]. Moreover, as accurately demonstrated by Suarez et al. [7], the overall mortality for these lesions is approximately 23%. However, when considering the lethality of the high-grade-only subset, this value increases to 70% or even 100% in cases with an accompanying glioblastoma component (Table 1) [8-13].
In their work, Schober et al. [9] were the first to tentatively assign a World Health Organization (WHO) grade to their diagnosis, while Cerda-Nicolas and Kepes [10] preferred to add a descriptive modifier (anaplastic). In a similar way, Sharma et al. [4] proposed to dichotomize these lesions into low-grade (benign) or high-grade (malignant/anaplastic) based on the Ki-67 proliferation index. In spite of these grading attempts, Snipes et al. [8] seminally pointed out that prognosis seems to be more related to location rather than to any morphological or immunohistochemical feature, though the degree of anaplasia and the mitotic index might be worthy of consideration. On the other hand, the case documented by Altamirano et al. [14] highlights the issue of whether atypia can exist in the fibroma portion of the neoplasm. This is exceedingly difficult to assert, as the boundaries of either a gliosarcoma or sarcoglioma are unavoidably blurred. In the aforementioned case, increased immunolabeling for Ki-67 was noted in the nuclei of the mesenchymal part, though it was rated in less than 5% of the overall tumour. The authors interpreted the mass as a proliferative activity of uncertain relevance. Though our case displays focal nuclear enlargement and vacuolization (Fig. 1D), we did not observe an increment in mesenchymal Ki-67 expression and did not find any areas suggestive of a true sarcomatous component. Nonetheless, this presumed “atypia” might not reliably worsen the prognosis, as in our case it was based on histology (glioblastoma) while in Altamirano’s group was determined by location (thalamus/mesencephalon). p53 immunostaining in our case was also of conjectural significance, indicating not only a possible secondary glioblastoma, but also a common origin of both the glial and mesenchymal elements, similar to the demonstration of Biernat regarding gliosarcomas [15], and thus possibly representing a true benign mesodermal differentiation as formerly hypothesised by Snipes et al. [8]. Furthermore, p53 immunolabeling has been previously tested in nine gliofibroma cases with no positive result [2,4,6,12,16-18]. Interestingly, Goyal et al. [19] also applied this immunohistochemical marker in three other cases but never mentioned the final outcome. This lack of p53 expression is shared by the so-called desmoplastic infantile astrocytoma and may hint, at least in these aforementioned cases, at a different developmental pathway from that of diffuse or anaplastic astrocytomas and glioblastomas.
Three more issues briefly deserve our attention. (1) As previously mentioned, the first report of gliofibroma clearly detailed a desmoplastic astrocytoma (Friede-type gliofibroma) [1]; nonetheless, this designation was subsequently applied to similar lesions consisting of both glial and mesenchymal derivatives (authentic glioma/fibroma) [10], as in the present case. It is worth mentioning that the glial constituent can range from innocuous to evidently malignant, while the latter is a consistently benign fusocellular component. (2) In the bimorphic subset, it has not been fully corroborated whether the two populations are distinct cell lineages or if the fusiform cells are glia capable of differentiating into fibroblasts, myofibroblasts, Schwann cells or histiocytes [2,20]. (3) Recently, some desmoplastic lesions displaying a non-astrocytic glial phenotype with immunohistochemical properties more akin to ependymoma (as in the case described by Gargano et al. [2]) have also been diagnosed as gliofibromas. Thus, as pointed out by Suárez et al. [7], a controversial dilemma of “splitting versus lumping” currently prevails in the conceptual and classification schemes regarding these kinds of tumours. Nevertheless, it seems to be more of an academic and histopathological predicament rather than one of clinical concern or prognostic relevance.
As shown in our case and previously advised by other authors, gliofibroma prognosis is greatly influenced by the degree of anaplasia of the glial component. Hence, we suggest adding a grade (WHO I–IV) or descriptive (low-grade, high-grade, anaplastic, malignant/glioblastomatoid) modifier to the diagnosis of gliofibroma.
It had been previously claimed that necrosis and prominent vascular proliferation are not usual features of gliofibromas [16]. Thus, we document the second case of gliofibroma with a glioblastomatous component [13] (though this is the first case involving a mixture of glioma and fibroma) as well as the third case occurring in Latin American population [2,14].
 XML Download
XML Download