

 PDF
PDF Citation
Citation Print
Print


Small round cell tumor (SRCT) is a collective term, not a single disease entity, that refers to a tumor composed of small, round and relatively undifferentiated primitive cells with scant cytoplasm under light microscopy after excluding neoplasms of the central nervous system (CNS) [1]. The term ‘small’ indicates slightly larger than or double the size of the red blood cells in air-dried smears [2]. Most of them are solid malignant tumors with histologic resemblance reflecting their dysembryonic, undifferentiated, and primitive features. Tumor cells of this category are poorly differentiated or undifferentiated with high nuclear/cytoplasmic (N/C) ratio and little cytoplasm, subsequently presenting an overall bluish picture on hematoxylin and eosin stain; thus, it is also referred to as small blue round cell tumor.[3] SRCT is actually a heterogeneous group of neoplasms dominating in childhood and adolescence, and histologic diagnoses of SRCTs are diverse, as shown in Table 1 [1,4-16]. Therefore, the diagnosis of SRCT is challenging, necessitating the use of multimodal ancillary techniques [17-19]. Traditional work flow for a diagnosis of SRCT was light microscopic examination with special stains at first and ultrastructural examination as a second step when the light microscopic findings are equivocal [4]. Poorly differentiated or undifferentiated tumors require further immunohistochemistry and cytogenetic molecular studies [19]. Currently, however, as a matter of convenience, immunohistochemistry has become the routine diagnostic modality due to the relative ease of use and interpretation, while electron microscopy (EM) is frequently skipped due to its declined usage in diagnostic fields [1,20]. Sometimes, cytogenetic or molecular studies are employed at firsthand because these methods are now widely considered as a confirmative modality in the diagnosis of SRCT. Not unexpectedly, a combination of more than two modalities results in a higher rate of specific diagnosis than either alone [4]. Immunohistochemistry frequently shows equivocally positive or negative reactions, particularly in poorly differentiated tumors, and overlapping of antigenicities between several SRCTs requires large panels of antibodies for the diagnosis [1,17,21].
Table 1.
Disease entities appearing as small round cell tumors along with ultrastructural differential characteristic findings
![]()
Over the last two decades, advances in molecular diagnostic techniques for SRCT or soft tissue tumors have been remarkable and such diagnostic techniques have been rapidly integrated. For example, the FLI1 protein, the gene product of FLI1, t(11;22), is detected in up to 85% of Ewing sarcoma/peripheral neuroectodermal tumors (ES/PNET), and WT1 on 11p13 is unique and central to the pathogenesis of desmoplastic small round cell tumor (DSRCT)[21-23]. It is yet uncertain whether SRCTs showing aberrant translocation should be included as a specific entity or be left as undifferentiated or high-grade sarcomas [13,24,25]. Although these cytogenetic or molecular studies are the most suitable for confirming a specific diagnosis and the usage of the “old” ultrastructural examination in the diagnostic field is still limited to some disease entities, the cytogenetic or molecular studies have several limitations [26]; requirement for very meticulous technique, discrepancy with histomorphology, low sensitivity, and unpredicted variant rearrangements.
As stated above, current usage of EM has declined as following limitation; first, EM lacks distinction between benignity and malignancy. Second, EM lacks heterogeneous findings due to varying differentiation and overlapping ultrastructural structures in different types of cells as well as sampling error [4,27]. Even with EM studies, some tumors still exhibit unknown lineage of differentiation, especially when the tumor cells are primitive and undifferentiated [28]. Although new models of digitalized EM have redeemed the time-consuming process and cost-ineffectiveness, EM has become unfamiliar to pathologists [12,29-31].
Here, we discuss the advantages and disadvantages of ultrastructural studies for the diagnosis of SRCTs with suggested applications of EM.
ULTRASTRUCTURAL FINDINGS OF SMALL ROUND CELL TUMORS
In this review, we explain SRCTs with following subdivisions; first, SRCTs are mainly comprised of tumors with unknown histogenesis. In this group, ES/PNET, neuroblastoma, DSRCT, and malignant rhabdoid tumor (MRT) are included. Second, certain types of soft tissue neoplasms can rarely appear as SRCTs. The third group is hematologic malignancies, including nonHodgkin’s lymphoma and leukemia [7,16]. Others include epithelioid gastrointestinal stromal tumor (GIST), small cell or rhabdoid type melanomas [8,15,32]. Among these groups, certain type of SRCTs can be diagnosed under light microscopy and immunohistochemistry by their unique histologic features such as GIST or small cell liposarcoma, which are not discussed here.
Go to : 
SRCT of unknown histogenesis
ES/PNET, neuroblastoma, DSRCT, and MRT are categorized as tumors of unknown histogenesis [8,31]. ES and PNET that present primarily in bone and soft tissues have been reported and previously studied as separate entities. However, there has been a recognition of a shared cytogenetic abnormality between the two and they are now regarded as different manifestations of the same entity at the genetic level (i.e., ES/PNET family tumors including Askin tumor) [12].
ES/PNET family tumors show diverse heterogeneous histologic features, including large cell atypical types or adamantinoma-like variant ES/PNET, which have shared moleculogenetic abnormalities. ES/PNET family tumors show varying degrees of neuroectodermal differentiation. The ultrastructural findings of ES/PNET show undifferentiated mesenchymal cells arising from a mesenchymal stem cell and differentiating to neural cells [12,33]. Ultrastructural findings of this neural differentiation include intermediate filaments, microtubules, or neurosecretory granules and processes [12]. Small amounts of glycogen can be seen in other types of tumors, but in ES, glycogen deposits usually form lakes or pools (Fig. 1A). The tumors show undifferentiated primitive cells with no discernible characteristics except for abundant glycogen pools. Rich glycogen within the cytoplasm is a notable and distinctive finding for ES/PNET, but is not specific [33]; MRT may show plentiful glycogen particles and no other specialized structures, mimicking ES/PNET (Fig. 1B) [11]. MRT of soft tissue show large paranuclear whorls of intermediate filaments, with entrapped organelles and lipid droplets [34]. These paranuclear cytoplasmic whorls of intermediate filaments are shared by DSRCT of the abdominal wall [5,21,22,35].
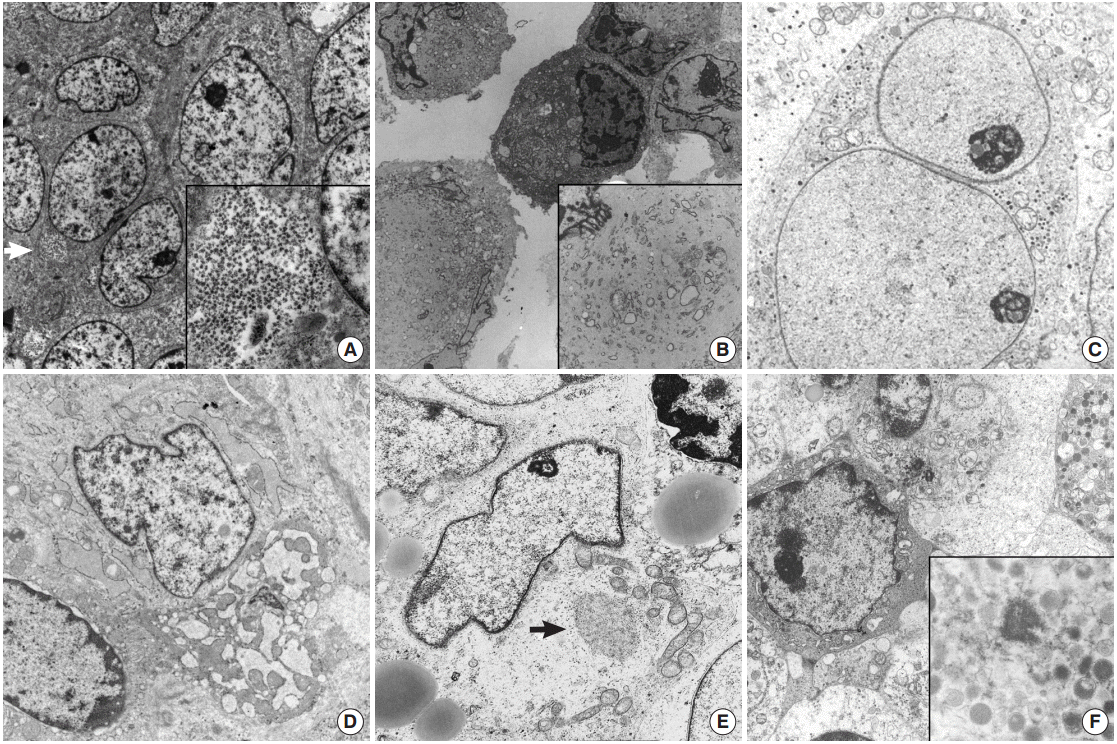 | Fig. 1.(A) Ewing sarcoma shows polygonal shaped cells having large round cells and a small amount of cytoplasm with mainly glycogen pools (arrow) (×7,500). Inset indicates abundant cytoplasmic glycogen particles (×10,000). (B) Tumor cells of malignant rhabdoid tumor show paranuclear whorls packed with intermediate filaments intermingled with cytoplasmic organelles (×2,000). Inset shows paranuclear whorls filled with dense intermediate filaments, dilated rough endoplasmic reticulum (RER) cisternae and degenerated mitochondria (×8,000). (C) Neuroblastoma. Round shaped tumor cells have neurosecretory dense core granules in the cytoplasm (×8,000). (D) Small cell osteosarcoma shows ovoid shaped tumor cells containing a moderate amount of mitochondria and ribosomes with dilated RERs (×v8,500). (E) Alveolar rhabdomyosarcoma, solid variant. Closely apposed round to ovoid shaped cells have irregular shaped nuclei and a moderate amount of cytoplasm containing lipid vacuoles, mitochondria, and Golgi apparatuses (×3,500). Arrow indicates myosin-ribosome complexes (×12,000). (F) Myeloid sarcoma demonstrates cytoplasmic myeloid granules, measuring 200 nm on average (×1,500). Inset shows membrane bound myeloid granules (×6,000).
|
DSRCT is a rare malignant pediatric tumor that usually affects the abdominal cavity [22]. When DSRCT is presented in visceral organs, its diagnosis poses significant difficulty. In such circumstances, DSRCT showing multidirectional differentiation may add diagnostic confusion [5,23,35]. Neural differentiation characteristics such as neurosecretory granules, microtubules, dendritic interdigitating processes, or epithelial differentiation such as cell junctions or intracytoplasmic lumen can also be identified in DSRCT. Confirmation of neurosecretory granules, formerly known as dense core granules, requires careful examination because they may be easily confused with primary lysosomes, which are larger than the former. DSRCT have mitochondria, lipid aggregates, and a few groups of paranuclear intermediate filaments [21]. The multidirectional differentiation is concordant with t(11;22)(p13;q12), resulting in a chimeric EWS/WT1 transcript [23]. Presence of paranuclear whorls of intermediate filaments corresponds to the dot-like immunostaining with desmin or vimentin, together with the absence of specialized ultrastructural structures [21]. Among pediatric SRCTs, a tumor showing only neural differentiation is a neuroblastoma, the most frequently encountered extracranial pediatric solid tumor [36,37]. It originates from neural crest cells of the sympathetic nervous system and EM is very helpful in its diagnosis. These neoplasms have neurosecretory granules (50–200 nm in diameter) with membranebound dense core granules, cytoplasmic processes containing parallel arranged microtubules of 20 nm in diameter, and primitive cell junctions (Fig. 1C). Cell junctions are not demonstrated in neuroblastoma.
Go to :
SRCT categorized as a soft tissue neoplasm
Soft tissue tumors are classified according to morphology and histogenesis. As up to 30% of soft tissue tumors show chromosomal alterations, integration of molecular study in the diagnosis of soft tissue tumors has been recommended. However, accumulating data on these molecular alterations sometimes further complicates the molecular diagnoses of some soft tissue tumors. Certain types of soft tissue tumors can present as SRCTs including monophasic synovial sarcoma [8], small cell osteosarcoma [18], mesenchymal chondrosarcoma [38,39], small cell variant of melanoma [15,40], solid variant of alveolar rhabdomyosarcoma [41], round cell liposarcoma [42], small cell schwannoma, and small cell malignant peripheral nerve sheath tumor (MPNST) [9,36]. ES/PNET family tumors are too undifferentiated to have characterized ultrastructural findings, and ultrastructural morphology is hardly distinguished from the undifferentiated cells of small osteosarcoma and mesenchymal chondrosarcoma of soft tissue [43-46]. In primitive cells differentiating to chondrocytes in mesenchymal chondrosarcoma, some thin cytoplasmic projections to the extracellular spaces are the only distinguishing finding [13,46-48]. The chondrocyte-like tumor cells usually found in conventional myxoid chondrosarcoma are not found in most undifferentiated forms of chondrosarcomas [48]. The chondrocytes of well differentiated forms are ovoid to fusiform shaped ones with an eccentric nucleus and cytoplasm having abundant mitochondria, conspicuous endoplasmic reticulum with dilated cisternae of rough endoplasmic reticulums (RERs), cytoplasmic glycogen, lipid droplets, and numerous thin cytoplasmic projections such as microvilli into the extracellular area, which is filled with glycosaminoglycan granules, and collagen fibrils [49]. The higher the grade of chondrosarcoma, the lower the number of organelles, along with less prominent cytoplasmic projections into the extracellular spaces, bizarre nuclei, and more prominent nucleoli [50,51]. More immature cells correspond to small undifferentiated cells mimicking fibroblasts. Cells of mesenchymal chondrosarcoma have rounded nuclei, scanty cytoplasm, and few organelles and glycogen, like ES/PNET [13]. The intercellular matrix is very scanty and does not contain collagen fibrils. The search for more differentiated foci and correlation with the light microscopic findings is rewarding in such cases. These well-formed microvilli are found in low to intermediate grade chondrosarcoma, and are well observed even in clear cell chondrosarcomas, which lack other organelles, but have abundant glycogen particles [51]. Small cell osteosarcoma should be differentiated from other more common SRCTs. Chondroblasts showing features similar to those of chondroblastoma or chondrosarcoma can be seen in osteosarcoma [18]. There are rare osteoblastic or multinucleated giant cells with a large amount of RERs without ruffled borders. The tumor cells of a rare variant of osteosarcoma are a primitive form of osteoblasts, and the critical point of its ultrastructural diagnosis is to search for extracellular osteoid, extracellular matrix containing collagen fibers with deposition of hydroxyapatite crystals, showing correlation with light microscopy. The tumor cells have round shaped nuclei with occasional infolding and a more moderate amount of cytoplasm than those of ES/PNET. They also have a considerable number of organelles, most of which are RERs and ribosomes (Fig. 1D). Although tumor cells in a small cell osteosarcoma resemble those of ES, osteoid will provide an ultrastructural distinguishing point as it does in the light microscopic diagnosis [17,52].
In pediatric age, a solid variant of alveolar rhabdomyosarcoma rarely presents as SRCT [41]. Like other tumors, the EM features of a solid variant of alveolar rhabdomyosarcoma depend on the degree of differentiation of rhabdomyoblasts [53]. Contrary to much more differentiated rhabdomyosarcoma, the primitive small cells are monomorphous round to elongated in shape and are surrounded by external lamina with a moderate amount of cytoplasm filled with glycogen and other organelles and have no easily detectable muscular features such as thick myosin-ribosome complexes or dense plaques. A careful search for thick myosin filament-ribosome complexes defined as 15-nm filaments intimately associated with numerous free ribosomes, the earliest evidence of rhabdomyoblast, can confirm the diagnosis of a rare solid variant of rhabdomyosarcoma (Fig. 1E) [41]. Ultrastructural evidence of skeletal differentiation such as specific myofilament arrays, focal density, Z-band material, and pinocytotic vesicles may not be detectable in solid variants of alveolar rhabdomyosarcoma [53]. Caution is needed because such neoplastic rhabdomyoblastic differentiation may be found in other SRCTs, such as malignant melanomas or schwannian Triton tumors.
MPNST can present with small cell components, and it may cause confusion in small biopsy samples [54]. For example, benign peripheral nerve sheath tumor such as schwannoma, may show small cell components [55]. The presence of small cell changes of peripheral nerve sheath tumors is mainly regarded as malignant transformation; however, it sometimes implies differentiation toward immature neural cells [9]. Distinguishing poorly differentiated small cell MPNST from ES/PNET is not easy, at least on histologic and immunohistochemical grounds alone because MPNST has neither a reliable immunohistochemical marker nor specific moleculogenetic changes. Therefore, EM is often useful in detecting elongated interdigitating cytoplasmic processes, pinocytotic vesicles, and fragments of basal lamina [56]. In particular, the presence of basal lamina around tumor cells under EM confirms bidirectional differentiation into melanocytes and nerve sheath tumor. In such cases, basal lamina can be found in epithelial, myoepithelial, endothelial, Schwann or perineurial cells, or muscle cells, but cannot be seen in neurons, glial, hematolymphoid cells, and myofibroblastic cells [57]. Therefore, misinterpretation of the basal lamina of these surrounding reactive cells for the lamina of tumor cells should be avoided.
Small cell variant of poorly differentiated monophasic synovial sarcoma, one of the poorly differentiated monophasic synovial sarcoma, should be distinguished from other SRCTs [8,58]. Monophasic synovial sarcoma showing epithelial membrane antigen–negative and cytokeratin-negative poorly differentiated one, requires either molecular cytogenetic study to evaluate the specific translocation t(X;18)(p11;q11) or EM examination [59]. Ultrastructurally, differentiation of synovial sarcoma ranges from unclassifiable spindle cells to fully differentiated epithelioid cells [60]. The spindle to fusiform-shaped tumor cells of synovial sarcoma has a high N/C ratio and non-descript cytoplasm devoid of schwannian differentiation, fibroblastic and leiomyosarcomatous differentiation, continuous basal lamina, long spacing collagen, and long intervening processes. Tapering bipolar processes joined by occasional rudimentary cell junctions or desmosomes, or infrequent tonofibrils must be distinguished from MPNST [60].
Go to :
SRCT of hematologic malignancies
The tumor cells of some SRCTs, particularly the solid variant of alveolar rhabdomyosarcomas, resemble lymphoblastic leukemic cells or non-Hodgkin’s lymphoma [61,62]. Conversely, dense infiltrates of relatively small undifferentiated tumor cells of uncertain origin, which mimic undifferentiated sarcoma, may be the first presentation of hematologic and lymphoid malignancies [63]. In particular, myeloid sarcomas with no evidence of circulating blasts may manifest as undifferentiated tumors with small round cell morphology, which frequently show aberrant immunophenotypes for myeloperoxidase, terminal deoxynucleotidyl transferase, CD68 (KP1), lysozyme, CD117, or CD34 [64]. Ultrastructural demonstration of myeloid granules of various morphology and stage are crucial in such cases [65-68]. Elongated Auer bodies as well as round or rod-like primary or azurophilic granules are useful ultrastructural findings. Primitive myeloid blasts have little cytoplasm but show greatly enlarged nucleoli with coarse granular nucleoplasm and thin peripheral heterochromatin (Fig. 1F) [16]. Myeloid tumors show morphological forms ranging from myeloblasts through promyelocytes to monocytes [68]. This is not the case for lymphoid malignancy [19,65,66] and subtyping of lymphoma has been attempted. EM cannot determine the line of differentiation in lymphoid malignancy, but it plays a role in excluding lymphoid malignancy. Abundant ribosomes and polysomes with an absence of cell junctions or basal lamina are the most common features of lymphoid malignancy.
Go to :
Other tumors presented with SRCTs
As previously mentioned in the SRCT of unknown histogenesis, MRTs showing rhabdoid morphology under light microscopy have been reported in every location of the body including the brain, liver, soft tissues, lung, skin, heart, and kidney, in which the tumor was originally described [11,69-72]. Recent immunohistochemistry reveals that the tumor cells have nuclear immunoreactivity for integrase interactor 1 (INI-1), which is critical for the diagnosis of this highly aggressive tumor, and show paranuclear bundles of intermediate filaments under ultrastructural examination [70]. Cell junctions are not observed in MRT. These paranuclear whorls of intermediate filaments are seen in other tumors of rhabdoid appearance; a rhabdoid variant of malignant melanoma also shows prominent nuclei and cytoplasmic accumulation of whorls of intermediate filaments, which compress the nuclei and a few organelles entrapped in the cytoplasm and melanosomes. As stated above, DSRCT also shares these ultrastructural findings. The so called rhabdoid appearance under light microscopy mimicking renal MRT (i.e., large cells with eccentrically located nuclei and abundant, eosinophilic cytoplasm) refers to tumors showing diffuse organoid sheets of round or polygonal shaped cells with paranuclear whorls of intermediate filaments. Tumors showing rhabdoid appearance are found in various tumors including pleomorphic fibrosarcoma with rhabdoid appearance, rhabdoid tumors of the CNS such as atypical teratoid/rhabdoid tumor, the CNS counterpart of MRT, rhabdoid meningioma, astroblastoma with rhabdoid appearance or rhabdoid choroid plexus carcinoma [70,71]. Extrarenal rhabdoid tumors are a heterogeneous group of neoplasms and it is also described as poorly differentiated neoplasms with rhabdoid features for these undifferentiated tumors [72]. Ultrastructural features shared with other tumors with rhabdoid features may not provide a diagnostic clue other than loss of INI-1 nuclear staining, which is critical for the diagnosis of this highly aggressive tumor.
Under light microscopy, small cell (Merkel-like) melanomas appearing in adolescents and children are composed of small undifferentiated cells of melanocytic-lineage [15]. Ultrastructural examination is required to determine if the cells are pre-melanosomes or melanosomes of various stages and differentiating them from the reactive cells is important [73]. Pathologists should search meticulously for melanosome granules of various stages.
Go to :
CONCLUSION
In summary, the current shift in SRCT diagnosis is towards immunohistochemistry and cytogenetic molecular techniques, but sometimes these techniques are inadequate. Even with careful traditional analyses, the correct diagnosis remains elusive. Immunohistochemistry has limitations in sensitivity and specificity, while moleculocytogenetic study has proven to be useful in characterization of SRCTs; however, it has also become evident that some genetic abnormalities are not tumor specific, and even demonstrate results not correlating with the histologic morphology. As stated above, light microscopy should remain as the main diagnostic tool. As EM has several overlapping features (Table 1), it may not be able to identify a line of differentiation if the tumor cells are cytologically undifferentiated. However, EM should be included in the standard diagnostic processes for SRCTs, as it can provide both validity and reliability in its specificity.
Here, we emphasize that pathologists need to understand the appropriate applications and pitfalls as well as the benefits of EM, which permits optimal diagnosis of SRCTs.
Go to :
 XML Download
XML Download