

 PDF
PDF Citation
Citation Print
Print



A mutation/deletion involving donor or acceptor sites for exon 14 results in splicing out of the exon 14 of the mesenchymal epithelial transition (MET) gene and is known as “MET exon 14 skipping” (∆MET14) [1-3]. This change in turn abolishes the site for ubiquitination and aborts the physiologically timed decay of MET protein [4]. Persistent MET activation turns it oncogenic. This genetic alteration has emerged as a Tier 1 biomarker [5] in non–small cell lung carcinoma (NSCLC) with a reported incidence of ~4% in lung adenocarcinoma [6,7] and 2% in squamous cell carcinoma [6]. This frequency supersedes that of other Tier1 driver alterations in NSCLC like ROS1 and BRAF p.V600E which occur at 1%–2% and 2%–3%, respectively [8]. The two recent approvals with substantial objective responses and improved progression-free survival to MET inhibitors, namely capmatinib [9] and tepotinib [10], necessitate the identification of this alteration upfront.
Next-generation sequencing (NGS), especially RNA sequencing, has emerged as the most relied platform to detect this variation as it detects the structural variation and nullifies the need to detect the 120 different mutations which may lead to this alteration [5]. DNA-based assay suffers from limitations in detecting large deletions (>20 bp) that are common at the 5' donor site [11]. Moreover, many popular NGS panels do not cover the 5' region of the exon 14 and the upstream region of the intron 13 adequately [10]. Far easier is the mRNA-based approach that simply demonstrates the fusion of exon 13 with exon 15 limiting the size and numbers of interrogated regions. However, the RNA-based NGS is not widely available. Besides, the lack of accessibility to NGS and design flaws in DNA panels that do not adequately cover all the mutational sites, necessitates a more practical alternative that can be used by basic molecular laboratories intending to provide ∆MET14 testing.
Single-gene assays by Sanger sequencing can be designed using DNA as a substrate. However, low analytical sensitivity and the lack of awareness of all genetic changes that leads to ∆MET14 limits the diagnostic utility of Sanger sequencing particularly in the context of limited availability of testing material.
We herein describe our experience of ∆MET14 detection by an mRNA-based assay using polymerase chain reaction followed by fragment sizing, with a final product size being 140 bp smaller in ∆MET14 compared to the wild type MET exon 14. The assay has been validated against an RNASeq NGS assay (Oncomine Focus Assay, Thermo Fisher Scientific, Waltham, MA, USA).
MATERIALS AND METHODS
Specimen collection
All cases of NSCLC that tested negative on single-gene testing for epidermal growth factor receptor (EGFR)/anaplastic lymphoma kinase (ALK)/ROS proto-oncogene 1 (ROS1) alterations and underwent NGS-based genomic profiling upfront at our center from 2018 to 2020 were included. The clinical and pathological features were retrieved and collated from the medical record archives of the hospital.
Next-generation sequencing
The NGS panel used was the Oncomine Focus Assay comprising 52 genes implicated across various solid organ malignancies, interrogating single nucleotide variations and fusions as well as copy number alterations. The details of this assay methodology can be found elsewhere [12].
Fragment sizing assay
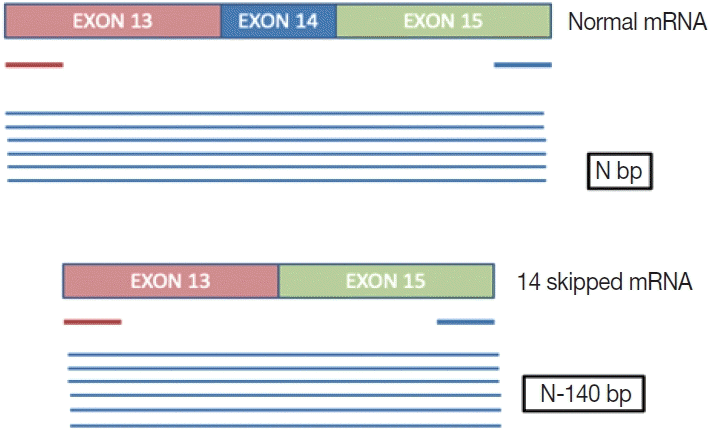
This is a home brew assay which was developed with the concept that the transcripts from true ∆MET14 will be shorter by ~140 bases than their wild type counterparts. The cases which were called MET exon 14 skipping positive on NGS were subjected to this assay, along with 13 healthy controls in order to establish the validity for true negatives (Fig. 1).
Primer design
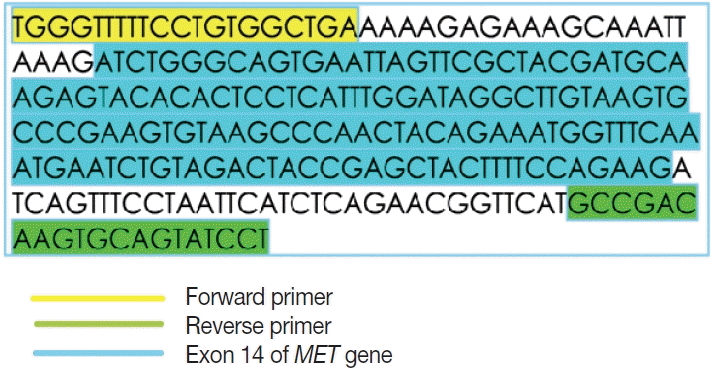
This is an RNA-based polymerase chain reaction (PCR) assay and the primers have been designed using hg19 human reference genome sequence of the MET gene (NM_001127500.2). The primers were designed using NCBI (National Center for Biotechnology Information) primer blast using cDNA molecule. The product size is 235 bp for MET exon 14 skipping wild type, whereas the MET exon 14 skipping mutant was 141 bp shorter. The primer design is depicted in Fig. 2 and the primer sequences are as follows: forward primer, 5'-TGGGTTTTTCCT GTGGCTGA-3'; reverse primer, 5'-AGGATACTGCACTT GTCGGC-3'.
To assess the presence of RNA, housekeeping gene β-actin is used as assay process and extraction control. The product size is ~92 bp which is concordant with the 94-bp product size of the mutant MET exon 14 skipping. The primer sequences used for β-actin are as follows: forward primer, 5'-CCACACTGTGC CCATCTACG-3'; reverse primer, 5'-AGGATCTTCATGAG GTAGTCAGTCAG-3'.
RNA extraction and cDNA synthesis
Total RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumor blocks using the Promega ReliaPrep FFPE Total RNA Mini-prep System (Z1002, Madison, WI, USA). Nucleic acid was quantified using Qubit 3.0 Fluorometer (Invitrogen, Life Technologies, Carlsbad, CA, USA). cDNA was synthesized from 10 ng of RNA using the SuperScript VILO cDNA synthesis kit (11754050, Thermo Fisher Scientific). The protocol followed was according to the vendor’s insert and the cycling conditions were as follows: reverse transcription step at 42°C for 30 minutes and enzyme denaturation at 85°C for 5 minutes.
Polymerase chain reaction for MET exon 14 skipping mutation
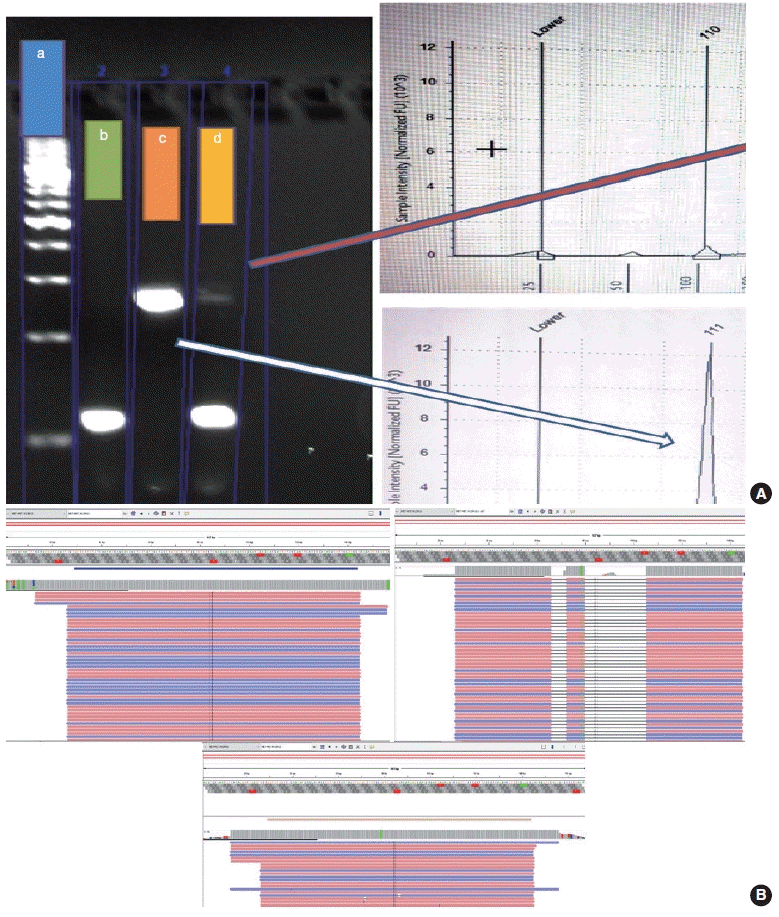
The PCR reaction mixture was prepared in proportions as per manufacturer recommendations. The Thermo Fisher Scientific Applied Biosystems Veriti 96-Well Thermal Cycler was used for amplifying this reaction mix with the following cycling conditions: 45 cycles of denaturation at 95°C for 30 seconds, followed by annealing at 56°C for 20 seconds, then elongation at 72°C for 30 seconds, and final extension step at 72°C for 7 minutes. The prepared PCR products were electrophoresed using 3% agarose gel. The expected PCR products using the primers described above are (1) MET exon 14 wild type, ~235 bp and (2) ∆MET14, ~94 bp. The presence of a single small band of ~94 bp was also considered positive for MET exon 14 skipping. For each reaction, a 100 bp ladder, a known positive control, and a known negative control along with no-template controls were run (Fig. 3).
Fragment length analysis using TapeStation
Following the analysis using agarose gel–based electrophoresis, the products were also analyzed using capillary electrophoresis/fragment length analysis on the Agilent 4200 TapeStation system (Agilent, Santa Clara, CA, USA). Tape station examination was done using high sensitivity D1000 DNA screen tapes for analyzing the size, quantity, and integrity of the samples. For each sample, 2 μL of high sensitivity D1000 sample buffer was added to 2 μL of the sample in the provided tube strip which was sealed using the cap strip. After this, the product was vortex mixed using IKA vortex at 2,000 rpm for 1 minute and spun at 1,000 rpm for 1 minute. The sample was then loaded in the TapeStation and analyzed using the Agilent TapeStation Analysis Software. The presence of a single peak at 235 bp length was considered MET exon 14 wildtype and the presence of single peak at 90–110 bp was considered mutant. The presence of two peaks, each at 235 bp and 90–110 bp, was also considered MET exon 14 mutant (Fig. 3).
RESULTS
Baseline characteristics
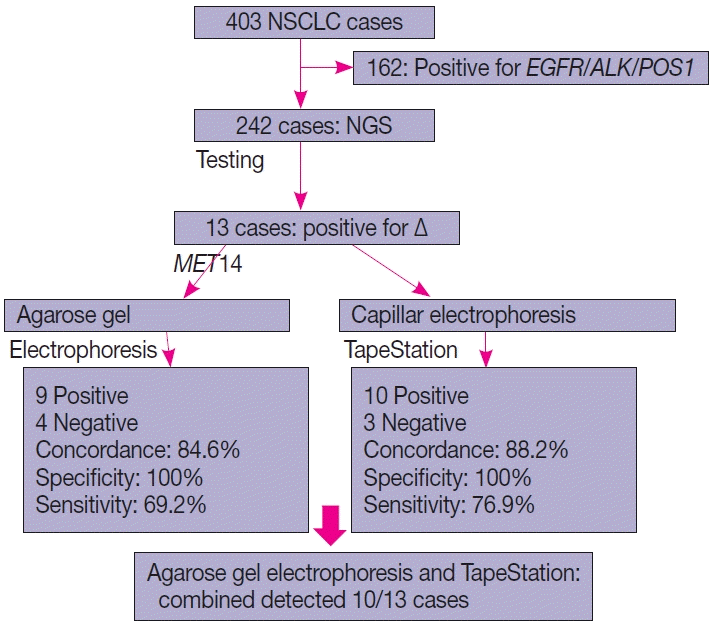
Of the four hundred and three cases of NSCLC, single-gene testing for EGFR mutations, ALK rearrangements, and ROS1 rearrangements were positive in 161 cases (40%) (EGFR alterations, 117 [29.1%] cases; ALK rearrangements, 32 cases [8%]; and ROS1 rearrangements, 12 cases [2.7%]). The 242 cases which were wild type for these three underwent NGS testing upfront, of which 13 cases of ∆MET14 (3.2%) were detected. The median age of the cohort was 61 years (range, 37 to 82 years). There were six males (46.2%) and seven females (53.8%). There were three smokers (23.1%) and 10 non-smokers (76.9%). All cases (100%) depicted adenocarcinoma histology with acinar pattern (Table 1).
Comparison of NGS and agarose gel electrophoresis
Thirteen cases of ∆MET14 mutation were detected on NGS using RNA-based sequencing. However, no corresponding DNA alteration in terms of indels or missense mutations was found in any of these cases. All these cases were subjected to agarose gel electrophoresis as described above, and nine cases were found to be positive on gel electrophoresis. Four of these cases were negative on gel electrophoresis. All 13 healthy controls also yielded a negative result. Considering the NGS as a gold standard, the overall concordance rate was 84.6%, with sensitivity of 69.2% and specificity of 100% (p<.01). The lack of any bands in the negative cases was not the consequence of poor quality of the extracted RNA, as the beta-actin housekeeper amplified and yielded an optimal product.
Comparison of NGS and automated electrophoresis using TapeStation
All 13 cases that were subjected to agarose gel electrophoresis also underwent automated capillary electrophoresis using TapeStation. 10 cases showed peaks at 95–100 bp (positive for ∆MET14) and three cases were negative. The overall concordance rate was 88.4% with a calculated sensitivity of 76.9% and specificity of 100% (p<.001). An overview of patient recruitment and results is depicted in Fig. 4.
DISCUSSION
∆MET14 has gained importance owing to the recent development and approvals of selective MET inhibitors like capmatinib and tepotinib [9,10]. These are usually reported at a frequency of 3%–5% in NSCLC [13], and in this study, the frequency was 5.3% among cases that were tested by NGS, whereas the frequency was 3.2% among the entire NSCLC cohort.
We described an mRNA-based fragment sizing assay using both gel and capillary electrophoresis and we demonstrated 100% specificity for both, with concordance rates of 84.6% and 88.2% with NGS, respectively. Currently, there are no single-gene assay kits available for the detection of this alteration. Several home brew assays have been described in literature using different modalities like real-time PCR and Sanger sequencing. Kim et al. [14] described the concordance of mRNA-based real-time reverse transcriptase PCR (qRT-PCR) and DNA-based Sanger sequencing against NGS as a gold standard, and demonstrated 100% sensitivity and 97.4% specificity for qRT-PCR. The same for Sanger-based assay was 61.5% and 100%, respectively. The specificity of our assay was also 100%; however, the sensitivity was lower than qRT-PCR, and higher than Sanger sequencing. This may be attributed to the inability to detect large deletions on DNA-based alterations, as well as lower sensitivity of Sanger sequencing. A similar assay has been described by O’Brien et al. [15] using a one-step RNA-based PCR assay to detect ∆MET14, and they reported that this assay could potentially detect MET exon 14 skipped RNA with a lower limit of detection of approximately 10%.
Cases that tested negative on fragment sizing assay and positive on NGS-based assay had low mutant allele frequency as observed on Integrative Genomics Viewer (IGV) (Fig. 3B). The NGS-based tests have a higher analytical sensitivity (limit of detection) of 1%, which is not likely to be matched by PCR-based assays as described here.
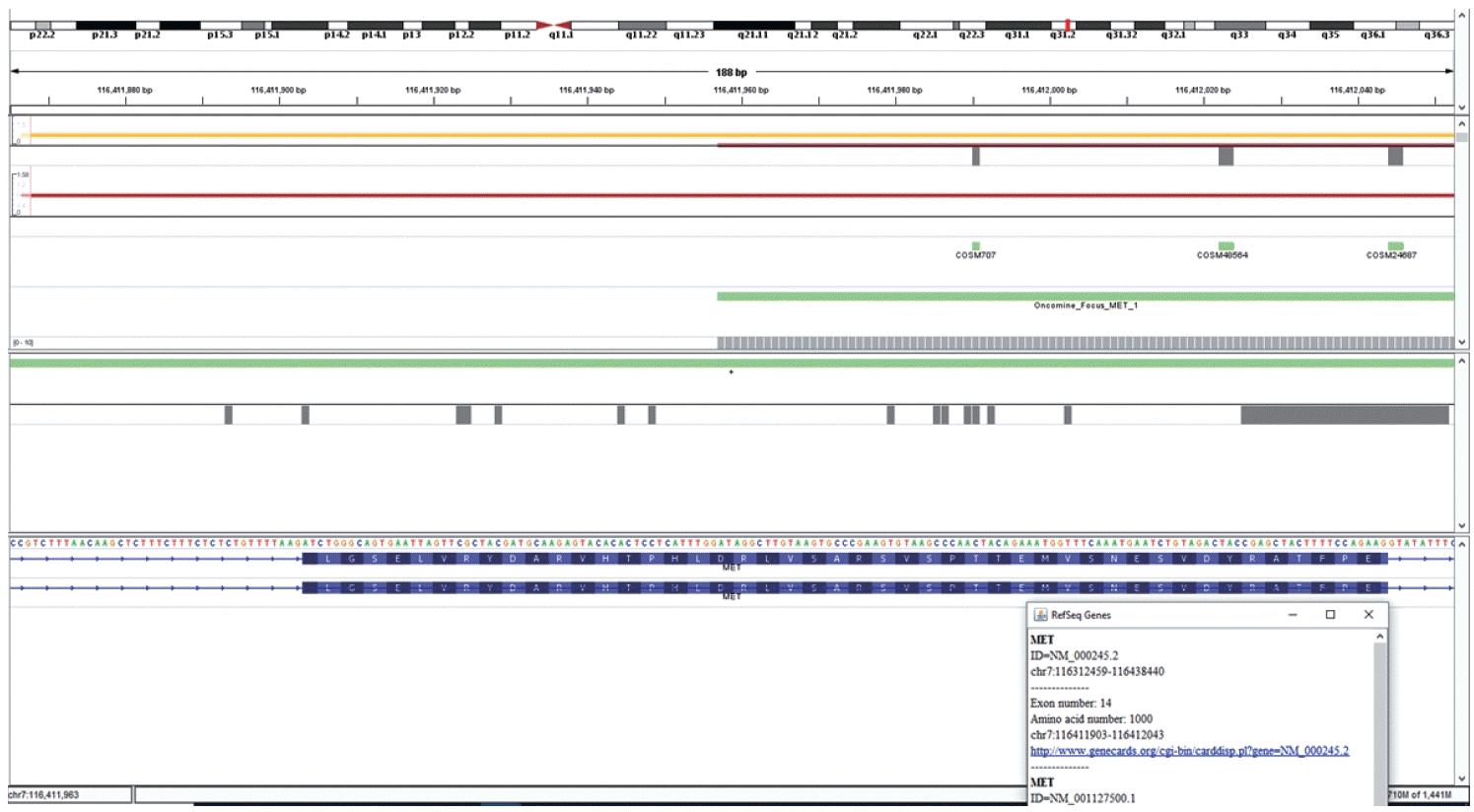
DNA-based alteration was not detected in any of the cases included in this study. This is attributed to the design of the Oncomine Focus Assay Panel. The 5' end of the exon 14 of the MET gene is not covered by the primers and amplicons are not generated for sequencing as depicted in Fig. 5. This region is the major site for missense variants and indels resulting in ∆MET14. These are therefore likely to be on DNA-based analysis.
There were four discordant cases on agarose gel electrophoresis and three on automated capillary electrophoresis. These are attributed to lower sensitivities of these two assays when compared to the NGS gold standard. However, combining both improved the sensitivity to 76.9%.
Although validated in-house, this assay suffers from a few limitations. The extraction process control used is beta-actin and not another conserved area of the MET gene, as most of the exons in the MET gene depict high rates of occurrence of single nucleotide polymorphisms. The assay has not been validated on an external control/cell line, and the samples used for validation are from known patients, hence the accuracy of the assay may not be representative. However, this assay will prove beneficial for small- and medium-sized labs where skilled technical personnel and NGS platforms are unavailable. Additionally, squamous carcinomas with ∆MET14 are also considered actionable and usually do not undergo broad panel-based testing, hence single-gene assays which are cost-effective will prove beneficial in such settings.
 XML Download
XML Download