

 PDF
PDF Citation
Citation Print
Print



Malignant rhabdoid tumor (MRT) of the kidney is an uncommon renal tumor mainly reported in children. This tumor is aggressive with a poor prognosis [1]. It was thought of as a tumor of the pediatric group until recently. Since the first reported case by Lowe et al. [2] in 1990, only limited case reports have been published on MRT of the kidney in adult patients [2-9].
Here, we report a rare case of MRT of the kidney in a 57-yearold male. The diagnosis was supported by immunohistochemistry and molecular studies. The microscopic, immunohistochemical, and molecular characteristics of this tumor are described in this report.
CASE REPORT
A 57-year-old Korean male presented with left flank pain lasting for a day at the urology department of an outside hospital. He was a previous smoker on medication for hypertension. He was diagnosed with spontaneous intracerebral hemorrhage 9 years ago and a ureter stone 7–8 years ago. Physical examination revealed left costovertebral angle tenderness. Under the impression of a ureter stone, a non-contrast computed tomography (CT) was performed, and two masses were found in the left kidney. Additional contrast-enhanced CT scans revealed a 7-cm-sized heterogeneously enhancing lobulated mass in the lower pole of the left kidney involving the Gerota’s fascia and a 4.4 cm-sized necrotic mass near the upper pole, raising suspicion for metastasis to a large necrotic lymph node in the hilum (Fig. 1A–C). The clinical diagnosis was renal cell carcinoma (RCC), stage IV. He was sent to our institution for surgical management, where the radiologists reviewed scans and drew the same conclusion (RCC, cT4N1) with some additional information: the mass might have invaded the left posterior abdominal wall, and the left adrenal gland abuts the upper pole mass. The patient underwent a radical nephrectomy with an adrenalectomy.
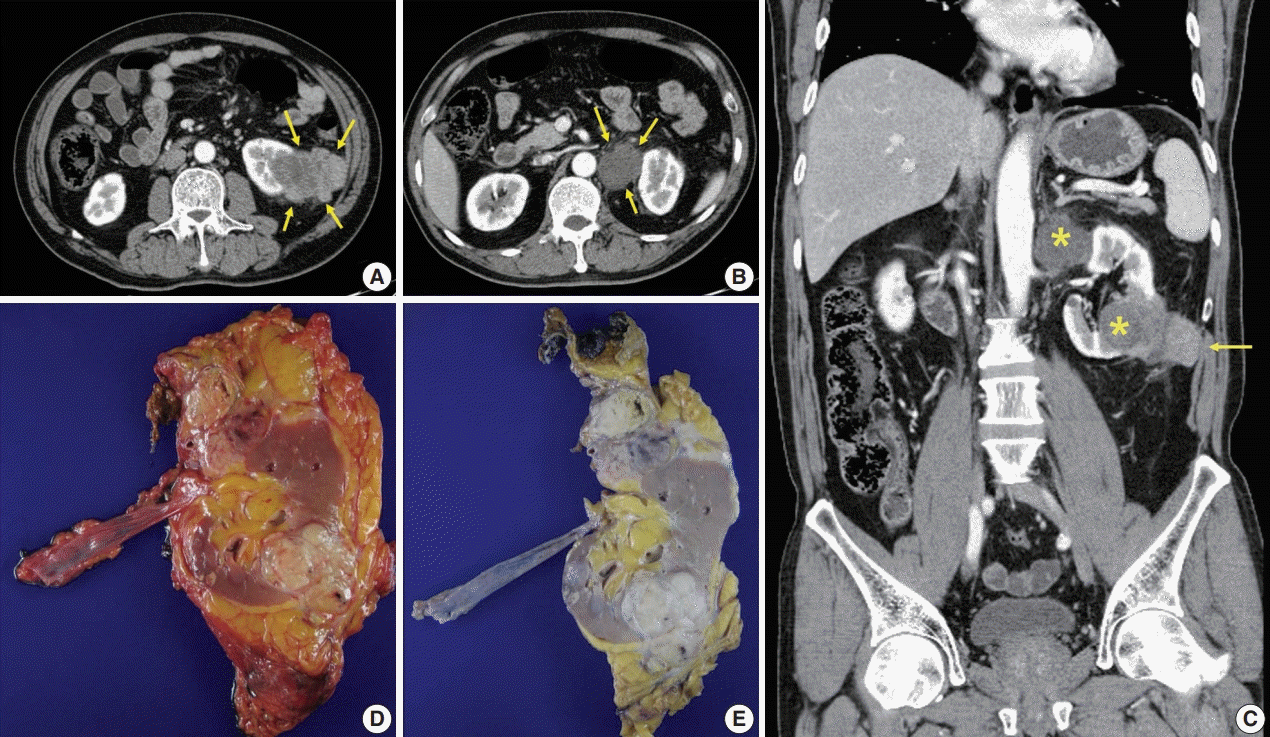 | Fig. 1.Radiologic and gross features of the tumor. (A) Axial abdominopelvic computed tomography (CT) scan showing a 6.5-cm-sized heterogeneously enhancing exophytic and lobulated mass (arrows) in the lower pole of the left kidney. Mild fat infiltration is seen around the mass, and invasion of the Gerota’s fascia is present. (B) Axial CT scan showing a 3.5-cm-sized mass (arrows) with homogeneously low density in the superior aspect of the left renal hilum. As the tumor compressed the kidney from the exterior, it was interpreted as a necrotic lymph node. (C) Coronally reformatted images of CT showing both masses (asterisks). The lower pole mass invaded the abdominal wall (arrow). (D, E) Surgical specimen before (D) and after (E) fixation. The kidney bore two well-circumscribed solid masses in the hilum near the upper pole and in the lower pole, measuring 4.5×3.5 cm and 7.0×4.1 cm, respectively. They were homogeneously yellowish-gray in color and firm with focal areas of hemorrhage and necrosis.
|
Specimens were then received for histopathological examination (Fig. 1D, E). The specimen consisting of the left kidney with perinephric fat, ureter, and adrenal gland measured 19×12×6 cm altogether. The kidney bore two well-circumscribed solid masses in the hilum near the upper pole and the lower pole, measuring 4.5×3.5 cm and 7.0×4.1 cm, respectively. On cross-sections, these two large masses were homogeneously yellowish-gray in color and firm, with focal areas of hemorrhage and necrosis predominantly located in the central portion of the mass. The lower pole mass extended to the pelvicalyceal system and the Gerota’s fascia. Renal vessels and the left adrenal gland were close to this tumor without direct contact. The hilar mass did not seem to arise from the renal parenchyma, but rather, it compressed the kidney from the outside. It was best considered a metastatic carcinoma to a lymph node.
Microscopically, the tumor was relatively well-defined, composed almost entirely of solid sheets of tumor cells, and separated by dense fibrous tissue at most interfaces. It had infiltrative borders on invasive fronts (Fig. 2A). Areas of necrosis were also present (Fig. 2B). Poorly cohesive, uniform round to polygonal epithelioid cells making solid sheets were seen in the main large mass on high power view. They had abundant densely eosinophilic cytoplasm with large eccentric nuclei, commonly known as rhabdoid features (Fig. 2C, D); nucleoli were prominent, and mitoses were frequently seen. Lymphovascular invasion was present. Apart from the main mass, multiple small round nodules with papillary architecture were also seen (Fig. 2E, F, H). A transitional zone between papillary nodules and the main mass was not present on slides. The papillary nodules were considered separate lesions consistent with small papillary adenomas.
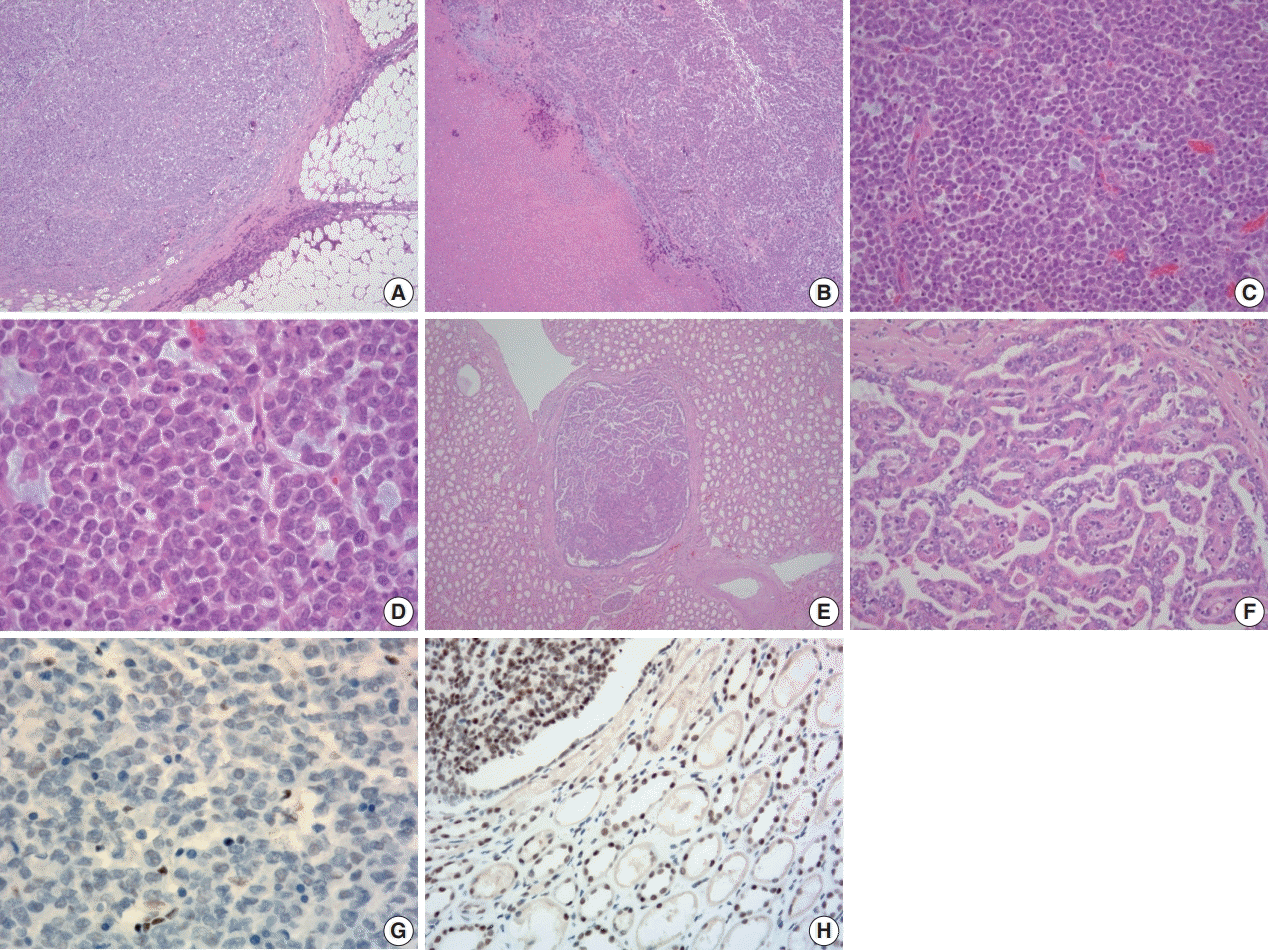 | Fig. 2.Histologic and immunohistochemical features of the tumor. (A) Microscopically, the tumor was relatively well-defined and separated by dense fibrous tissue at most interfaces. (B) Areas of necrosis were present. (C) The tumor was composed almost entirely of solid sheets of tumor cells. (D) High power view revealing poorly cohesive uniform round to polygonal epithelioid cells. They had abundant densely eosinophilic cytoplasm with occasional round eosinophilic inclusion and large eccentric nuclei with irregular borders. Nucleoli were prominent. (E, F) Multiple small round nodules with papillary architecture were observed irrespective of the main mass. They were positive for cytokeratin 7 (not shown) and interpreted as papillary adenoma. (G) Tumor cells showing loss of immunoreactivity for integrase interactor 1 (INI-1; 1:100 dilution). (H) Retained immunoreactivity for INI-1 is shown in the normal renal tubular epithelium and papillary area (upper left) (1:100 dilution).
|
Immunohistochemical staining for multiple antigens was done to diagnose this tumor with uncommon morphology. Results are shown in Table 1. Tumor cells were immunohistochemically positive for CD10 and focally positive for epithelial membrane antigen, pan-cytokeratin, and vimentin, but negative for cytokeratin 7 and CD117. They were also negative for immune cell markers (such as leukocyte common antigen, CD3, CD20, CD138, and CD68) and muscular markers such as actin and desmin. The tumor cells were negative for HMB45 and CD99, ruling out the possibility of primitive neuroectodermal tumor/Ewing’s sarcoma. Nuclear p53 was focally positive. After intra- and inter-departmental consultation, a diagnosis was made. The pathologic diagnosis for the large main mass in the lower pole at the time was RCC, unclassified with rhabdoid differentiation.
Table 1.
Results of immunohistochemical staining for tumor cells
![]()
A year after the surgery, his follow-up CT scans showed a newly visible 6.5-cm-sized irregularly lobulated mass with heterogeneous enhancement abutting the left psoas muscle underlying the left nephrectomy site (Fig. 3A). This was considered a regional recurrence invading the left psoas muscle and the posterior aortic wall. After two months, the mass had increased in size. It also extended to the right aortic wall. Another operation to reduce the burden of this newly-developed mass was performed. The specimen sent for examination was a conglomeration of lymph nodes measuring 7.5×6×3.5 cm in size and 86 g in weight (Fig. 3B). Cross-sections revealed whitish-tan solid and firm cut surfaces with focal necrosis and hemorrhage (Fig. 3C). Microscopic findings were very similar to the previously resected unclassified RCC, showing solid sheets of rhabdoid cells effacing lymph nodes diffusely and infiltrating capsules to form a conglomerated mass (Fig. 3D–F). The pathologic diagnosis was metastatic carcinoma from the unclassified RCC with pericapsular soft tissue extension and necrosis.
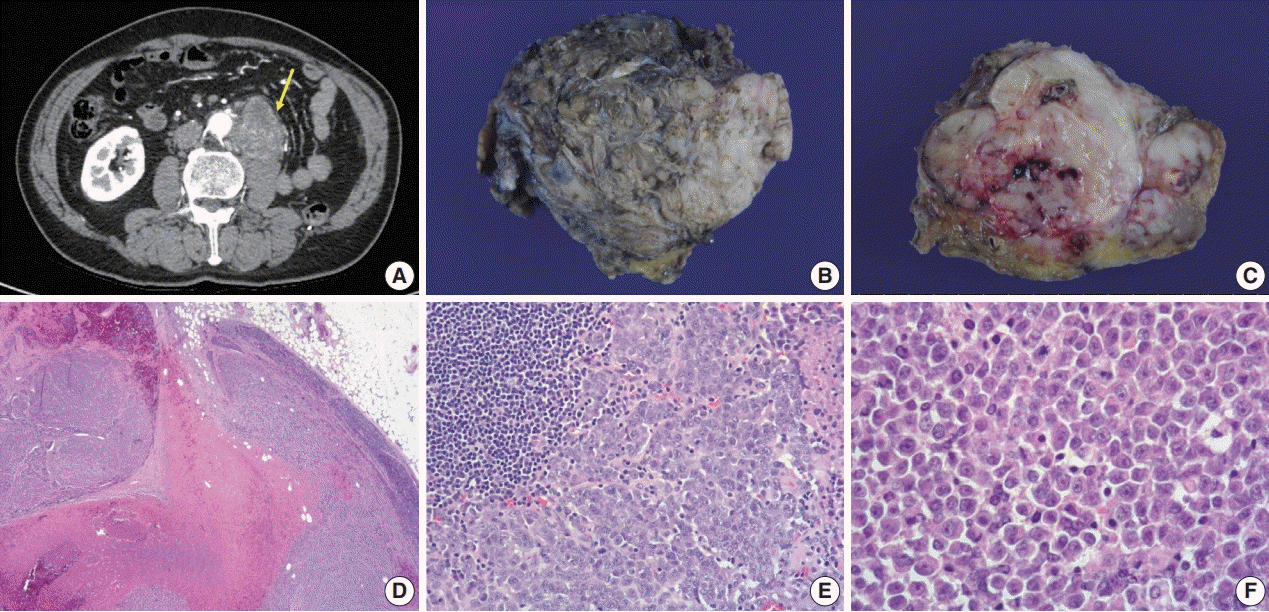 | Fig. 3.Radiologic, gross, and histologic features of the newly-developed mass. (A) Follow-up axial abdominopelvic computed tomography scan a year after the surgery, revealing a newly visible 6.5-cm-sized heterogeneous enhancing lobulated mass (arrow) in the anterior aspect of the left psoas muscle underlying the left nephrectomy site, extending to the posterior aspect of the infrarenal abdominal aorta. Thus, aortic invasion was suspected. It was considered a recurrent tumor in the lymph nodes. (B) The specimen sent for examination was a conglomeration of lymph nodes (7.5×6×3.5 cm). (C) Whitish-tan solid and firm cut surfaces with areas of focal necrosis and hemorrhage were seen. (D) Solid sheets of tumor cells diffusely effacing lymph nodes and area of necrosis were seen. (E) Lymphoid tissue (upper left) and poorly cohesive round to polygonal tumor cells are shown. (F) Tumor cells had eosinophilic cytoplasm with occasional round eosinophilic inclusion and large eccentric nuclei with prominent nucleoli, resembling the former tumor from the kidney.
|
To treat the remnant mass after the surgery, we referred the patient to the oncology department, where he was scheduled for palliative chemotherapy with temsirolimus. Infusions were given every 7 days for 4 weeks, which constituted one cycle. However, after the first infusion of the 4th cycle, he was unable to continue therapy due to the deterioration of his general condition. Upon cessation, the tumor was aggravated. It had spread to the level IV lymph node of the neck and was pathologically confirmed as metastatic carcinoma. Unfortunately, after three weeks of palliative radiation therapy, the patient died.
In the present case, immunohistochemical staining for INI1 and genetic/molecular analysis was not performed at the time of diagnosis due to technical limitations. However, several studies have shown that specific molecular aberration involving switch/sucrose non-fermentable–related, matrix-associated, actin-dependent regulator of chromatin, subfamily B member 1 (SMARCB1, hSNF5/INI1) on chromosome 22, which can be identified by a lack of immunohistochemical staining with INI1, is an biomarker for malignant rhabdoid tumors [10]. Therefore, additional immunohistochemical staining and next-generation sequencing (NGS) were retrospectively performed to rule out the possibility of a malignant rhabdoid tumor.
We used the Axen cancer panel for NGS that included 535 SNV/InDel genes and 54 fusion genes. NGS revealed a novel mutation in the SMARCB1 gene on chromosome 22 (c. 1047G>A, p.Trp349*). We further performed additional NGS for both normal and tumor tissues using a customized panel including 88 genes developed by our laboratory. The same result was obtained. Allele frequency was 81% in the tumor tissue and 49% in the normal tissue. The method for NGS was as follows. DNA was quantified using a Qubit 2.0 (Life Technologies, Burlington, Canada). Hybridization-based enrichment libraries were prepared using a SureSelectXT Target Enrichment System for Illumina Paired-End Sequencing Library (Agilent Technologies, Palo Alto, CA, USA), following the manufacturer’s protocol. Briefly, 200 ng of genomic DNA was fragmented, denatured, and hybridized with capture oligos during library preparation for highthroughput sequencing. Captured sequences were then enriched with streptavidin-conjugated paramagnetic beads and further amplified before being subjected to Illumina sequencing. Fragment sizes for all libraries were measured using a 2100 Bioanalyzer (Agilent Technologies). Then, qPCR was performed with a LightCycler 480 System (Roche Diagnostics, Indianapolis, IN, USA) using a Kapa library quantification kit (KK4854, KAPA Biosystems, Wilmington, MA, USA). Sequencing was performed using an Illumina NextSeq500 platform with an average read length of 2×150 bp per the manufacturer’s instructions.
Tumor cells showed a loss of INI1 expression (Fig. 2G). It is known that renal medullary carcinoma can also be entirely solid with sheet-like architecture or with exclusively rhabdoid morphology. It has also been reported that renal medullary carcinoma with rhabdoid features shows the absence of INI1 expression [11]. However, the tumor showed no immunohistochemical staining for PAX8 and PAX2, suggesting it was not from a renal epithelial origin. From a retrospective point of view, all morphologic, immunohistochemical, and molecular findings were sufficient for the diagnosis of MRT of the kidney. Our revised diagnosis of this tumor was MRT of the kidney. The bad prognosis of this patient was compatible with that seen in this tumor group.
Go to : 
DISCUSSION
NGS identified a novel nonsense mutation, c.1047G>A in exon 8/9 of the SMARCB1 gene, caused by p.Trp349*. This mutation halted the translation of the INI1 protein, a hallmark of MRT. Rhabdoid cells were first described by Gokden et al. [12] in the adult kidney. Rhabdoid cells have been described as variably cohesive epithelioid cells with eccentrically located vesicular nuclei with prominent nucleoli. Large intracytoplasmic eosinophilic hyaline globules are their characteristics. They resemble rhabdomyoblasts but differ from rhabdomyoblasts in that they do not show rhabdomyoblastic differentiation immunohistochemically or ultrastructurally [12]. MRT occurs mainly in the kidney of children less than 1 year of age. It is rare and has an aggressive clinical course [1]. Renal MRT was originally described as a ‘‘rhabdomyosarcomatoid variant of Wilms tumor’’ by Beckwith and Palmer [13] in 1978. However, after Versteege et al. [14] demonstrated potentially inactivating mutations or deletions of both alleles of the INI1 gene in almost all renal and pure extrarenal MRTs, they were regarded as a distinct entity with a specific molecular pathogenesis. The deletion of 22q11.2 and the mutation of hSNF5/INI1, encoding a chromatin-remodeling protein with the potential to alter global transcription patterns, are strongly associated with the development of a rhabdoid tumor phenotype [14,15].
One unique aspect of the present case was the presentation of a rhabdoid tumor of the kidney in an adult. Until recently, only a few cases have been reported on a pure rhabdoid tumor of the kidney in adults. In one study, germline mutation of hSNF5/INI1 in humans and its deletion in knock-out mice resulted in rhabdoid tumors developing at an early age [15]. Shannon et al. [16] have mentioned that this might be why pure rhabdoid tumors of the kidney are seldom reported in adults. Their study found identical VHL mutations in both clear cell and rhabdoid components of composite tumors in two patients. They argued that some rhabdoid tumors might arise from clear cell RCCs. They also mentioned the possibility of a sporadic biallelic inactivation of the gene(s) involved in the development of a rhabdoid phenotype taking place in genetically unstable tumor cells of a preexisting neoplasm [16]. If that is the case, a continuum might exist between an RCC with rhabdoid features and MRT of the kidney. As molecular and genetic studies were not done for the rhabdoid tumor cases of the aforementioned study, immunohistochemical staining for INI1 and genetic testing for the SMARCB1/INI1 gene should be performed for more cases of RCCs with rhabdoid features and malignant rhabdoid tumors of the kidney of adults to prove the diagnosis.
To obtain papers on MRT of the adult kidney, we searched for previously published case reports in PubMed. Among the results, we found eight case reports of pure MRT of the native kidney arising in an adult patient that was written in English [2-9]. Clinicopathologic characteristics of the cases are summarized in Tables 2 and 3 [2-9]. Age at diagnosis ranged from 32 to 79 years, and sex predominance was not noted. Two cases occurred in Asians, and five cases were presented in Caucasians. The race of the patient in the remaining case report was not recorded. Their initial symptoms were either related to the mass effect or metastasis. The maximal dimension of the mass varied from 3.5 to 12 cm. All cases except three underwent radical nephrectomy. Most of them showed bad outcomes. One case with pulmonary metastasis was treated with interleukin-2 (IL-2) therapy and showed an exceptionally better outcome than others. In another case, chemotherapy, including IL-2, failed [4,5]. Common pathologic findings were as follows. First, they showed solid sheets of non-cohesive cells with eccentric nuclei and large round eosinophilic cytoplasmic inclusion-like structures. Second, for cases that included an assessment of ultrastructure, large cytoplasmic whorls of intermediate filaments were present. Third, tumor cells were consistently immunohistochemically reactive for vimentin, either focally or diffusely, while they variably expressed cytokeratin and epithelial membrane antigen. These morphological, immunohistochemical, and clinical findings were consistent with previously described characteristics of MRT of the kidney by Wick et al. [17]. However, molecular or genetic investigation concerning the SMARCB1 gene was not performed for any of the patients. Immunohistochemical staining for INI1 was performed for only one patient.
Table 2.
Clinical features of malignant rhabdoid tumors of the kidney in adults reported in the literature
| Study | Year | Age/Race | Sex | Initial symptom | Size (cm) | Location | Management | Outcome | Medical history |
|---|---|---|---|---|---|---|---|---|---|
| Lowe et al. [2] | 1990 | 32/Caucasian | F | Right flank pain (3 days) | 12 | Rt. lower | Radical nephrectomy | Multiple metastases (3 mo after the diagnosis), declined further therapy | Meningomyelocele, amniotic bands, hydronephrosis |
| Clausen et al. [3] | 1994 | NA/Caucasian | F | NA | NA | NA | NA | NA | NA |
| Ebbinghaus et al. [4] | 1995 | 56/Caucasian | M | Persistent, non-productive cough (6 mo) | 6 × 9 | Lt. | Floxuridine (5 mo), interferon-alpha (3 mo), IL-2 (2 discontinued due to angina pectoris) | Good performance (over 18 mo from the diagnosis and 9 months after IL-2 therapy cessation) | CABG due to atherosclerotic coronary artery disease (10 yr) |
| Peng et al. [5] | 2003 | 38/Caucasian | F | Left flank pain (10 days) | 8 × 5 | Lt. upper to mid | Radical nephrectomy, several courses of chemotherapy including IL-2 | Chemotherapy: failed (died 5 mo after surgery) | - |
| Zhao et al. [6] | 2013 | 59/Asian (Chinese) | M | Weight loss of 6 kg (several months) | 5 × 4 × 5 | Lt. lower | Radical nephrectomy | A 10 mo postoperative follow up revealed no indications of tumor recurrence or metastasis | - |
| Podduturi et al. [7] | 2014 | 60/Caucasian | M | Progressive fatigue and abdominal distension (6 mo), worsening dyspnea (2 wk) | 3.5 × 2.5 × 2 | Lt. mid to lower | Radical nephrectomy, retroperitoneal lymph node dissection | Died during reoperation | Hypothyroidism, hypertension, chronic obstructive pulmonary disease |
| Okumura et al. [8] | 2019 | 79/Asian (Japanese) | F | Pain in the right buttock and difficulty in ambulation | 6.4 x 4.8 | Lt. upper | Treated with axitinib | Died 5 mo after the first hospital visit (3.5 mo after the treatment initiation) | Cerebral infarction at the age of (15 yr) |
| Ayari et al. [9] | 2019 | 65/NA | M | Right flank pain with progressive fatigue, a weight loss of 5 kg, abdominal distention (1 mo) | 5.6 x 5.3 | Rt. | None (died before therapeutic decision) | Died few days after the diagnosis | Hypertension, renal calculi, Lt. nephrectomy for hydronephrotic kidney (20 yr) |
![]()
Table 3.
Pathologic features of malignant rhabdoid tumors of the kidney in adults reported in the literature
| Study | Year | Gross feature | Extension/Metastasis | IHC |
|---|---|---|---|---|
| Lowe et al. [2] | 1990 | Poorly demarcated, solid, hemorrhagic | Extended beyond Gerota’s fascia densely adherent to the duodenum and the colon | Keratin (AE1/AE3), vimentin: (+) |
| EMA: focally (+) | ||||
| Desmin, myoglobin, muscle-type actin: (–) | ||||
| Clausen et al. [3] | 1994 | NA | NA | NA |
| Ebbinghaus et al. [4] | 1995 | Only biopsy done | Multiple metastasis to bilateral lungs | Vimentin: focally (+) |
| Desmin, S-100 protein or HMB-45 (–) | ||||
| CK: weak (+) | ||||
| Peng et al. [5] | 2003 | White to grayish tan and flesh, focal areas of necrosis | Invaded renal pelvis, calyces, the perihilar and perirenal adipose tissue, extended to the capsule, enlarged hilar lymph node | Vimentin: diffuse (+) |
| EMA, NSE, S-100: focal (+) | ||||
| p53: focal nuclear (+) | ||||
| Keratin (AE1/AE3, CAM5.2, CK7, and CK20): (–) | ||||
| CEA, SMA, myoglobin, light chains, HMB-45, MART-1: (–) | ||||
| Mucin: (–) | ||||
| Lymphoma markers: (–) | ||||
| Ki-67 (MIB-1): > 95% | ||||
| Zhao et al. [6] | 2013 | White to gray with a fleshy texture, focal areas of necrosis and hemorrhage | Focal invasion into capsule, retroperitoneal and left costophrenic angle lymph node metastases, metastasis to left lung | Vimentin: diffusely (+) |
| NSE, S-100, EMA: focally (+) | ||||
| Pancytokeratin, CK7, myoglobin, desmin, MSA, and SMA: (–) | ||||
| Podduturi et al. [7] | 2014 | Poorly circumscribed, soft tan lesion, numerous tan-gray nodules within perinephric and renal sinus | Extended from the hilum to the cortex, metastasis to bilateral lungs | Vimentin, pan-CK: strong (+) |
| Desmin, myoglobin, S-100, melanoma cocktail, TTF-1, CK7, CK20, CDX2, PAX8, CD31, CD34, factor VIII, CD30, CD45: (–) | ||||
| INI-1: (–) | ||||
| Okumura et al. [8] | 2019 | Only biopsy done | Multiple metastatic bone and lymph node lesions | Vimentin, EMA, CAM5.2, p53: (+) |
| CK, CD10: focally (+) | ||||
| INI1, CK7, CK20, AMACR, S100, CD45, RCC marker, ALK, α-SMA, desmin, MyoD, myogenin, HMB45, melan A: (–) | ||||
| Ayari et al. [9] | 2019 | Only biopsy done | Voluminous lymph nodes along the paraaortic region, compressing the inferior vena cava | Vimentin: strongly (+) |
| Myoglobin, desmin, SMA: (–) |
IHC, immunohistochemistry; EMA, epithelial membrane antigen; NA, not applicable; HMB-45, human melanoma black 45; CK, cytokeratin; NSE, neuron-specific enolase; CEA, carcinoembryonic antigen; SMA, smooth muscle actin; MSA, muscle specific actin; TTF-1, thyroid transcription factor-1; INI-1, integrase interactor 1; AMACR, α-methylacyl-CoA racemase; RCC, renal cell carcinoma; ALK, anaplastic lymphoma kinase;
![]()
Pediatric rhabdoid tumor of the kidney has been regarded as a distinct neoplasm, whereas rhabdoid differentiation in adult RCC is usually found in association with conventional (clear cell) tumors, from which it is thought to evolve [16]. In a retrospective analysis of 480 renal tumors conducted by Gokden et al. [12], 23 RCCs with rhabdoid features were found, all of which had at least 50% of the non-rhabdoid carcinoma components described as conventional (clear cell) RCC. The rhabdoid component was assumed to have arisen from the conventional RCC area [12]. Shannon et al. [16], on the other hand, reported 5 RCCs with rhabdoid differentiation, with three of them being composite tumors and two of them having rhabdoid elements only (which they called pure rhabdoid tumors). Several authors have described that loss of INI1 is helpful to differentiate MRT from other composite tumors, including primary renal neoplasms with rhabdoid differentiation where the expression of INI1 is retained [12,18,19].
Consistent with the role of SMARCB1 as a tumor suppressor gene, tumors can arise following the inactivation of both SMARCB1 alleles. While the majority of tumors are histologically classified as rhabdoid tumors, the loss of SMARCB1 expression has also been observed in other tumors, including epithelioid sarcoma, renal medullary carcinoma, undifferentiated pediatric sarcomas, and a subset of hepatoblastomas. It has been previously estimated that 15% to 20% of all rhabdoid tumors are caused by germline mutations of SMARCB1.
Although the present case’s initial diagnosis was undifferentiated RCC with rhabdoid features, there was a difference between this case and previously published cases of RCC with rhabdoid features. Unlike previously reported cases, the present case did not have any area with features of conventional RCC. There was no transition zone between the undifferentiated rhabdoid area and the classical RCC area, usually seen in RCC with rhabdoid features [12]. In practice, however, the rhabdoid area of RCC in adults is frequently viewed as a dedifferentiated area rather than raising the possibility for a diagnosis of malignant rhabdoid RCC. Indeed, some authors in the past have argued that pure extrarenal MRT in older patients might merely be poorly differentiated neoplasms with pseudo-rhabdoid features [20]. In addition, MRT of the kidney has been reported only in a limited number of adults at present, while more reports have discussed RCC with rhabdoid features. Furthermore, a small RCC component might have been missed during sampling. These altogether led to a faulty diagnosis of undifferentiated RCC with rhabdoid features.
In the present case, all morphologic, immunohistochemical, and molecular findings supported an MRT of the kidney. To the best of our knowledge, the present case is the first reported case of MRT of the native kidney in an adult, demonstrating both the loss of INI1 expression and the mutation of the SMARCB1 gene. This case points out again that MRT should no longer be considered merely a childhood tumor entity but rather a tumor that can also develop in adults. As a result, when an undifferentiated area with rhabdoid morphology is found in an adult tumor, one should initially look for areas showing classical morphological features of tumors that commonly arise in the affected organ. If these features are found, the transition zone between the two areas would be helpful to determine whether the rhabdoid area is the dedifferentiated area of the original tumor. If not found, additional sampling of the tumor with thorough inspection might be needed. When it comes to the conclusion that the tumor consists entirely of rhabdoid cells, immunohistochemical staining for INI1 and molecular or genetic investigation of the SMARCB1/INI1 gene on chromosome 22 could be helpful to diagnose an MRT in an adult.
Go to :
 XML Download
XML Download