

 PDF
PDF Citation
Citation Print
Print



Cancers arising from the ampulla of Vater, though designated as one entity of ampullary cancer (AC), are heterogeneous at morphological and molecular levels as well as in clinical behavior. The ampulla is a common channel formed by joining of the common bile duct and pancreatic duct that opens onto the intestinal (INT) surface, involving cancers arising from biliary, pancreatic ductal, or INT epithelium. These cancers exhibit complex morphological and molecular characteristics and can be located within the ampulla, be exophytic on the INT surface as a papillary lesion, or present as an infiltrative/stricturous lesion involving the ampullary bulb [1]. AC is a rare cancer and accounts for less than 1% of all gastrointestinal malignancies [2,3]. INT and pancreatobiliary (PB) differentiation exhibit distinct relationships with prognosis, where PB behaves aggressively compared to the INT phenotype [4]. The two differentiation types respond differently to chemotherapy [5–7]. The genomic biology and behavior of AC from India has not been well-studied. The genetic landscape of AC is evolving and has not been fully elucidated. In this study, we present the molecular profile of AC and the spectrum of genetic alterations in INT and PB differentiation.
MATERIALS AND METHODS
Fresh tumor tissues from 38 consecutive cases of AC and paired blood samples as normal controls were included for whole-exome sequencing (WES). Fresh specimens of pancreaticoduodenectomy were received in the pathology department, where the tumor content was estimated on frozen sections, and 3–5 mm tumor tissue in its greatest dimension was stored at −80°C before processing for WES. Following this, the specimen was fixed in 10% buffered formalin and processed for routine histopathology.
Histopathology
All the cases were histologically evaluated for tumor grade, depth of infiltration (pT category, American Joint Committee on Cancer 8th edition), lymphovascular invasion, perineural invasion (PNI), lymph node metastasis, and distant metastasis. The differentiation of tumors was categorized as INT, PB, or mixed according to the criteria of Kimura et al. [8] and later modified by Albores-Saveedra et al. [9]. The INT differentiation was similar to that of colorectal cancers with glands lined by tall columnar epithelial cells having elongated basally located nuclei frequently displaying pseudo-stratification and occasional apical mucin. The PB differentiation resembled pancreatic cancer with simple small glands and cuboidal to low columnar epithelium, markedly pleomorphic nuclei with or without nuclear pseudostratification, and intense desmoplastic reactions. Cases with > 30% of both subtypes were classified as mixed differentiation. Immunohistochemistry (IHC) for differentiation (INT: CDX2, cytokeratin [CK] 20, MUC2; PB: CK7, CK17, MUC1) was used in cases with mixed histological differentiation, which then were re-categorized into INT and PB subtypes. Those with equivocal IHC results were retained as mixed differentiation which histologically required any one of the differentiation types to be > 30%, but < 70% of the overall tumor area [10].
Whole-exome sequencing
DNA was extracted from fresh tissue and blood using a Qiagen GeneRead FFPE kit (Qiagen, Hilden, Germany), and the quality was assessed on QIAXPERT and Cubit. The Agilent Sure Select XT Kit (V5 + UTR) (Agilent Technologies, Santa Clara, CA, USA) was used for library preparation, and sequencing was performed on the HiSeq 2500 Ilumina platform (Illumina, San Diego, CA, USA) with a mean coverage of 100 × for blood DNA and 200 × for tumor tissue DNA.
Variant analysis
The bioinformatics tools used for alignment were BWA, GATK, SAM, and PICARD. For variant calling and annotation, Strelka, VariMAT, and OncoMD were used. Variant effect prediction was analyzed using Polyphen2, SIFT, Mutation Assessor, CONDEL, PhyloP, FATHMM, and SiPhy.
Functional, structural, and pathway analysis
Functional, structural, and pathway analyses of the mutated genes were studied with Gene Ontology software using the PANTHER (Protein Analysis Through Evolutionary Relationships) classification system and Reactome ver. 72.
Validation of the five most frequent mutations was performed by real-time polymerase chain reaction (PCR) and deep targeted sequencing.
RESULTS
Thirty-eight cases of AC were analyzed by WES. One case was excluded from the study because the blood DNA failed quality control before library preparation. The rest of the 37 cases that were successfully sequenced and analyzed for WES were composed of 26 males and 11 females with an age range of 24–76 years (mean, 53.4 years; median, 54 years).
Histological differentiation was categorized as INT (Fig. 1A, B) in 21 cases and PB (Fig. 2A, B) in 12 cases, along with mixed differentiation in four cases according to the morphological criteria. IHC exhibited expression of INT markers (CDX2, CK20, and MUC2) (Fig. 3) in one case and PB markers (CK7, CK17, and MUC1) (Fig. 4) in another case. Two cases of mixed differentiation with overlapping expression of IHC markers were kept in the mixed category. Therefore, with the combined morphology and IHC panel, 22 cases were classified as INT, 13 cases as PB, and two cases as mixed differentiation. The correlations of different histological parameters with INT and PB differentiation are shown in Table 1. Lower tumor pathological (T-) category was significantly associated with INT differentiation, and high PNI was associated with PB differentiation.
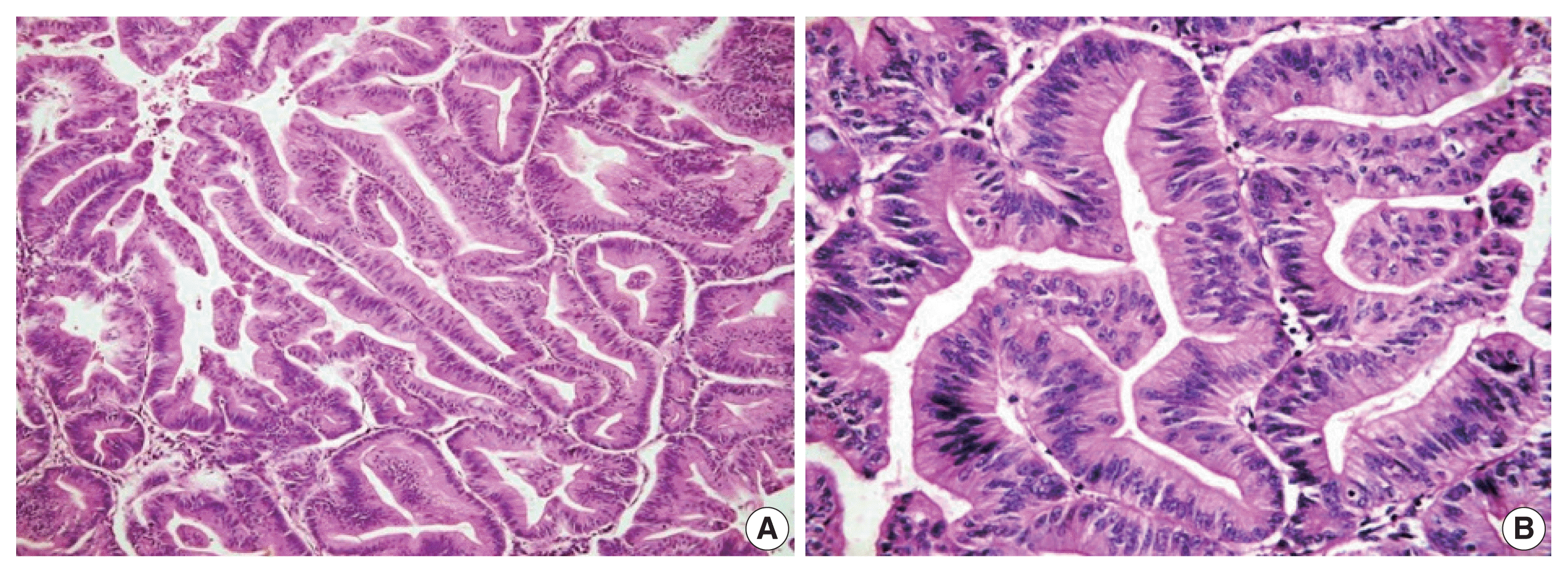
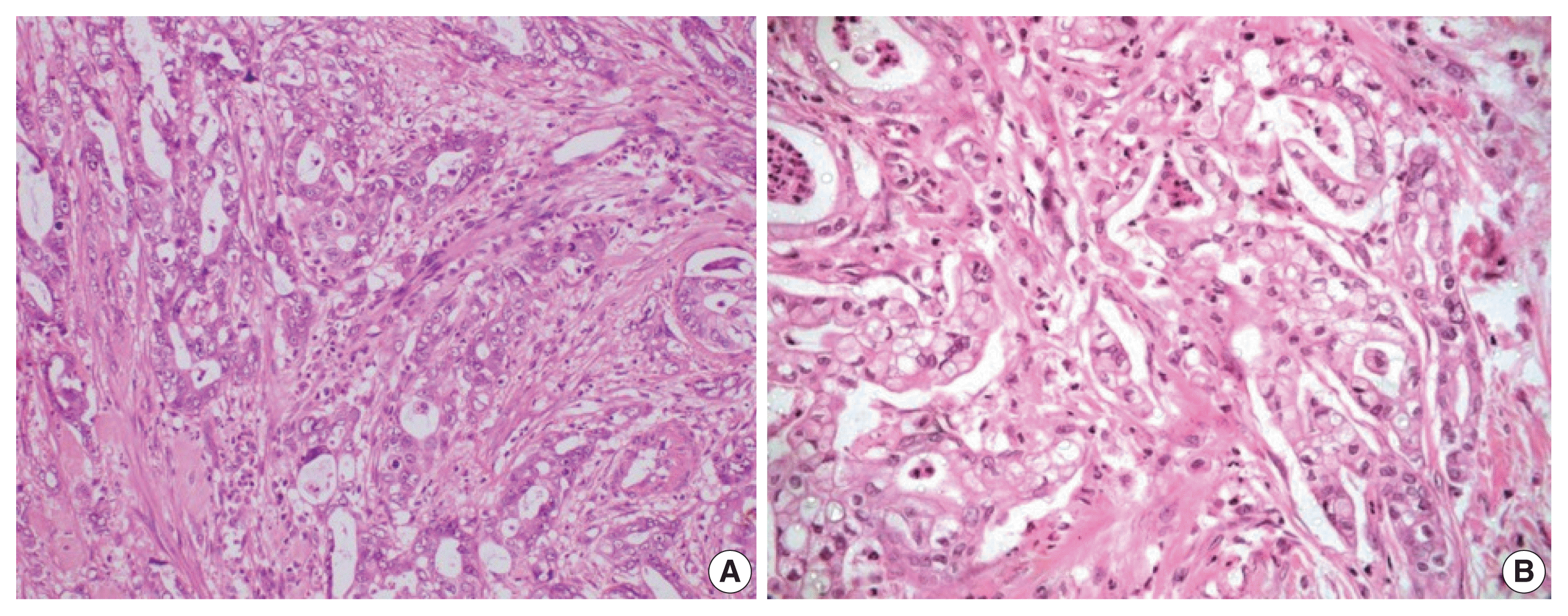
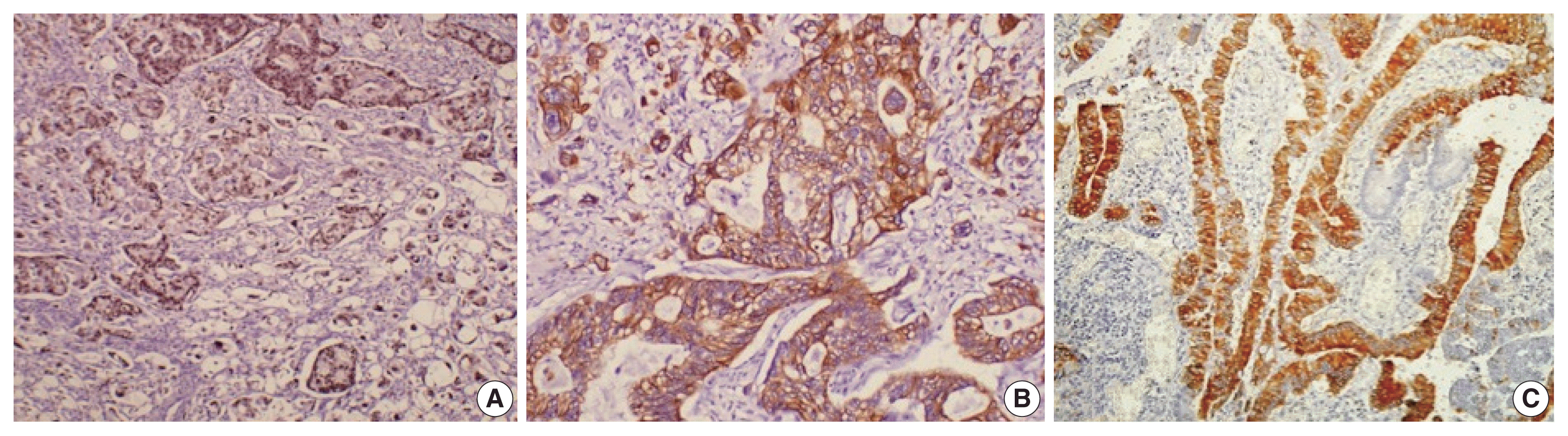
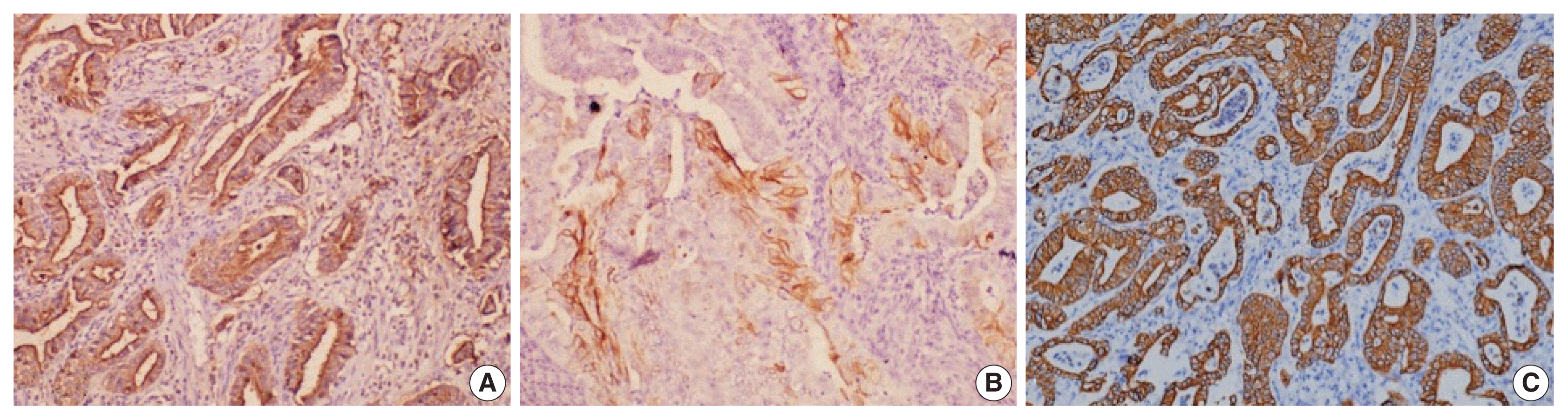
Fig. 1
Microphotograph of intestinal differentiation. Low-power (A) and high-power (B) views showing tall columnar cells with elongated to oblong basal nuclei and nuclear stratification.
![]()
Fig. 2
Microphotograph of pancreatobiliary differentiation. Low-power (A) and high-power (B) views showing cuboidal to low columnar cells with rounded centrally placed nuclei with no nuclear stratification. Desmoplastic stroma can be observed between the tumor glands.
![]()
Fig. 3
Immunohistochemical stains (A, CDX2; B, cytokeratin 20; C, MUC2) expressed in the intestinal type of ampullary cancer.
![]()
Fig. 4
Immunohistochemical stains (A, MUC1; B, cytokeratin [CK] 17; C, CK7) expressed in the pancreatobiliary type of ampullary cancer.
![]()
Table 1
Histological characteristics of ampullary carcinoma according to histological differentiation
![]()
Whole-exome analysis
The overall read alignment was approximately 99.97%, and the average passed alignment (percentage of reads aligning to hg19) was 97% for all samples. The average on-target coverage was approximately 85.67%. Read depth for each variant ranged between 10–560 (mean coverage 100 ×) in blood DNA and between 1–378 (mean coverage 200 ×) for tumor DNA. The variants present in blood were subtracted from the tumor tissue variants to exclude the germline variants present in these 37 patients. Variants present only in the coding regions were included in further analysis (Fig. 5).
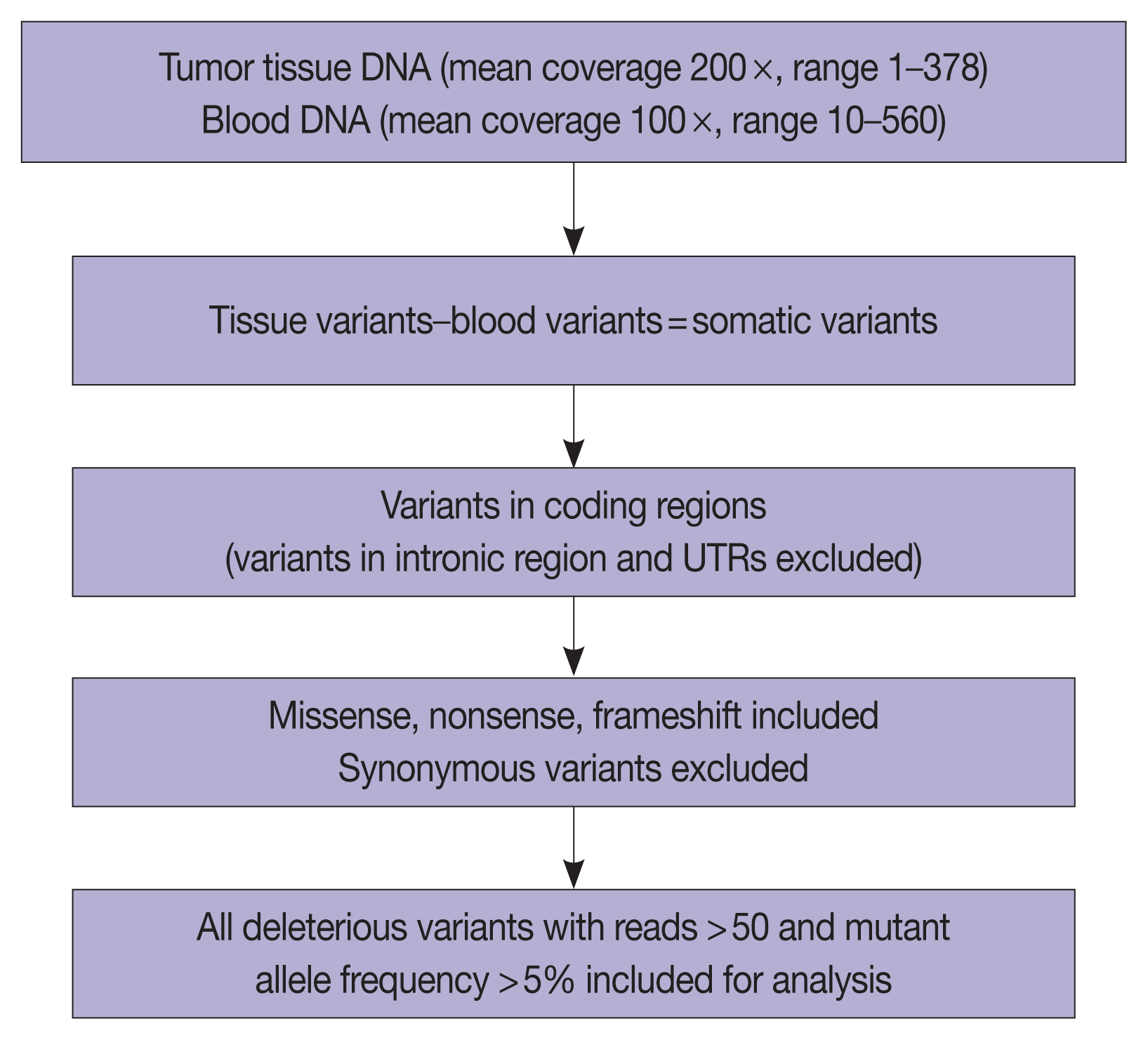
The number of total somatic variants in tumor DNA ranged from 14 to 30,153. Somatic variants having fewer than 50 reads and a mutant allele frequency <5% were excluded from the analysis, after which 1,263 somatic variants were observed, with 2 to 257 variants per patient. Furthermore, after excluding non-deleterious variants, there were 183 variants across 112 genes, with 78 exhibiting variations in INT and 51 in PB differentiation (Supplementary Table S1). Five cases (four cases with PB and one case with INT differentiation) did not show any deleterious somatic variant. In the 32 remaining patients, deleterious variants ranged from 1 to 31 per patient in patients with INT differentiation and 1 to 29 per patient in patients with PB differentiation (Fig. 6A, B). In the two cases that had mixed differentiation, both showed mutations in KRAS, while mutations in SMAD4, KEAP1, STK11, and CSFR1 were found in either of the two cases (Supplementary Table S2). Five of the genes that showed mutations in at least 10% or more of patients (APC, 20.5%; CTNNB1, 10.2%; KRAS, 25.6%; SMAD4, 20.5%; TP53, 33.3%) were validated by real-time PCR and/or targeted next-generation sequencing. The WES revealed targetable mutations in genes involved in one or more major pathways in cancer in 86.5% of all patients with AC of both INT and PB subtypes. KMT2 complex, APC, SMAD4, EPHA complex, ERBB complex, and Notch complex of genes were mutated at a higher frequency in INT types of AC compared to PB types, in that decreasing order, even though the difference was not statistically significant (Table 2). TP53 and KRAS were mutated nearly equally in INT and PB differentiation.
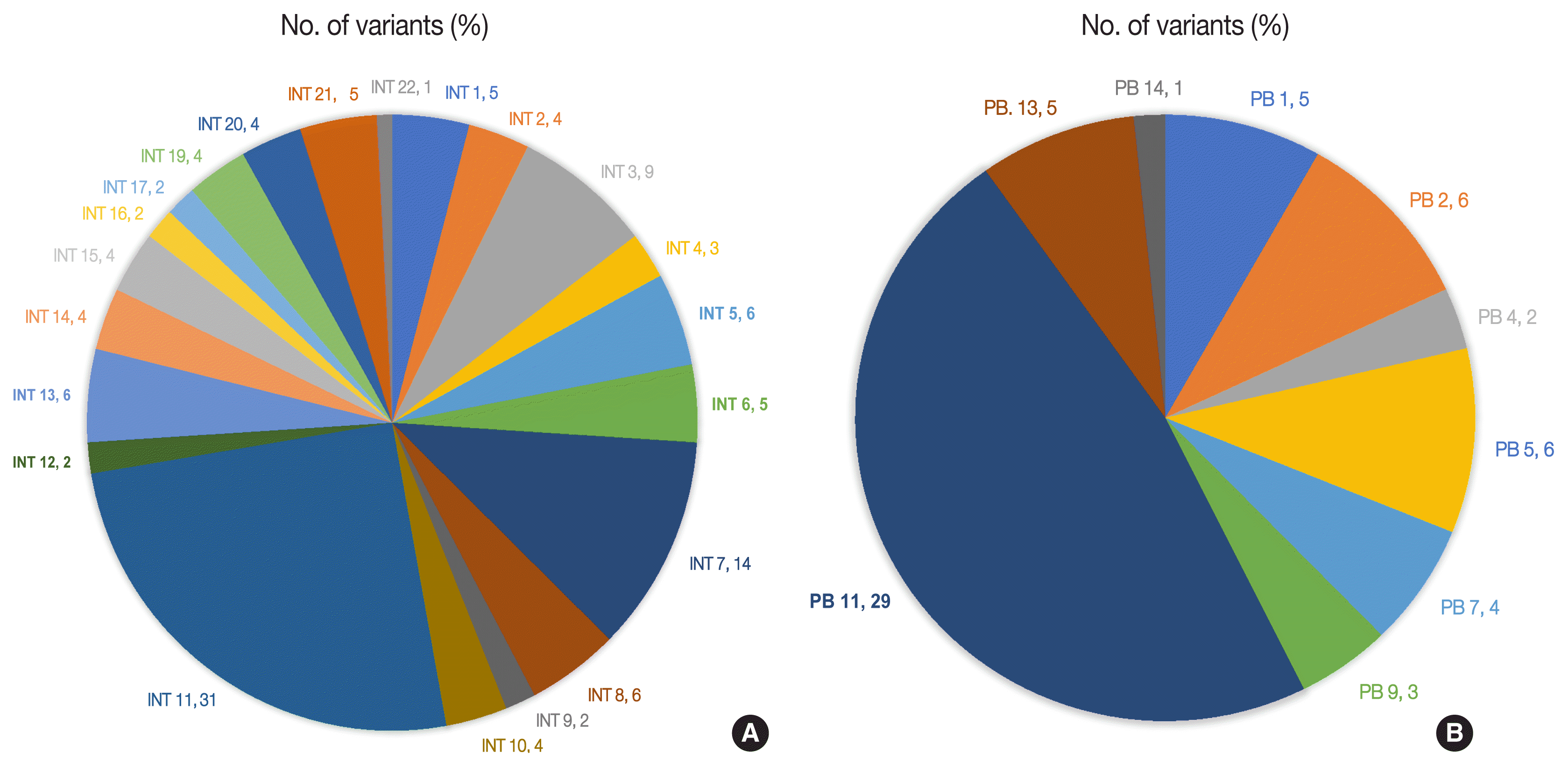
Fig. 6
(A) Deleterious somatic variants in intestinal (INT) ampullary cancers. (B) Deleterious somatic variants in pancreatobiliary (PB) ampullary cancers.
![]()
Table 2
Most commonly mutated targetable genes in patients with intestinal or pancreatobiliary differentiation
![]()
Functional, structural, and pathway analyses of variants in INT and PB subtypes
Functional, structural, and pathway analyses of the mutated genes were compared between INT and PB subtypes of AC. The number of genes responsible for different cellular components such as organelles and protein-containing complexes were mutated more frequently in INT than PB subtype, whereas it was similar for both subtypes for genes coding membrane and cell junctions (Fig. 7A).
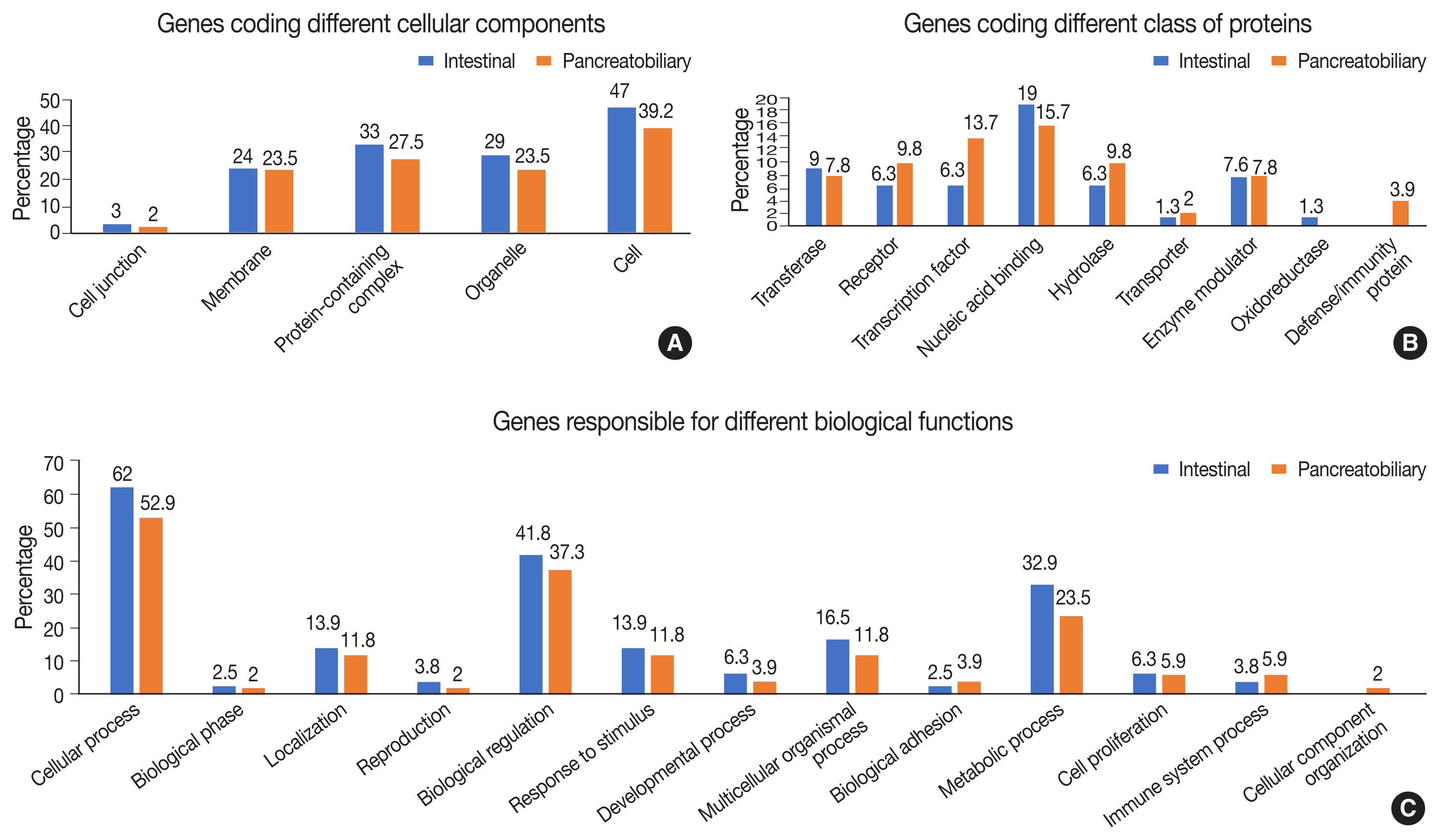
Fig. 7
(A) Genes forming part of the cellular component in intestinal and pancreatobiliary types of ampullary carcinoma. (B) Genes responsible for different classes of proteins in intestinal and pancreatobiliary types of ampullary carcinoma. (C) Genes involved in different biological functions and processes in intestinal and pancreatobiliary types of ampullary carcinoma.
![]()
In the group of genes coding for different classes of proteins, mutations were frequent in the INT subtype compared to the PB subtype, whereas the genes responsible for transcription factors, receptors, and hydrolases were mutated more frequently in the PB subtype (Fig. 7B). The gene coding proteins involved in defense and immunity were mutated in the PB subtype. In relation to different biological functions and processes, mutations in genes coding cellular and metabolic processes and biological regulation were slightly higher in the INT subtype than the PB subtype. Mutations in genes responsible for transcription regulation were significantly more frequent in the PB subtype, while those for binding and catalytic activity were marginally higher in the INT subtype (Fig. 7C).
Analysis of the signaling pathways revealed 31 signaling cascades that were significantly mutated (p > .002) in AC (Fig. 8). We divided the signaling pathways into six major groups of TP53, RAS–RAF–mitogen-activated protein kinase (MAPK), PI3-AKT, WNT, transforming growth factor β (TGF-β), and chromatin remodeling pathways, similar to Yachida et al. [11] and Gingras et al. [12]. The combined RAS-RAF-MAPK/PI3-AKT pathway was most frequently altered, in 70% of AC with ~82% in INT differentiation and 46% in PB differentiation (p = .021). This was followed by TP53 (~38%), WNT signaling (~32%), TGF-β (24%), and the chromatin remodeling complex pathway (16%). The PI3-AKT pathway was mutated predominantly in the INT type (63.6%), while the RAS-RAF-MAPK pathway was most frequently mutated in PB differentiation (59.1%). The next most common pathways to be mutated in the INT type were RAS-RAF-MAPK in 59.1%, WNT in 45.4%, TP53 in 41.0%, TGF-β in 22.7%, and the chromatin remodeling complex pathway in 13.6% of cases. On the other hand, PB differentiation harbored mutations predominantly in the RAS-RAF-MAPK pathway (46.2%), followed by TP53 (38.4%), PI3-AKT (30.7%), TGF-β (22.7%), chromatin remodeling complex (23.1%), and the WNT pathway (15.4%). The results suggest that PI3-AKT, RAS-RAF-MAPK, and WNT cascades were more frequently mutated in INT type, while the chromatin remodeling complex was more frequently mutated in the PB type. TP53 and TGF-β pathway alterations were similar in the differentiation types (Table 3).
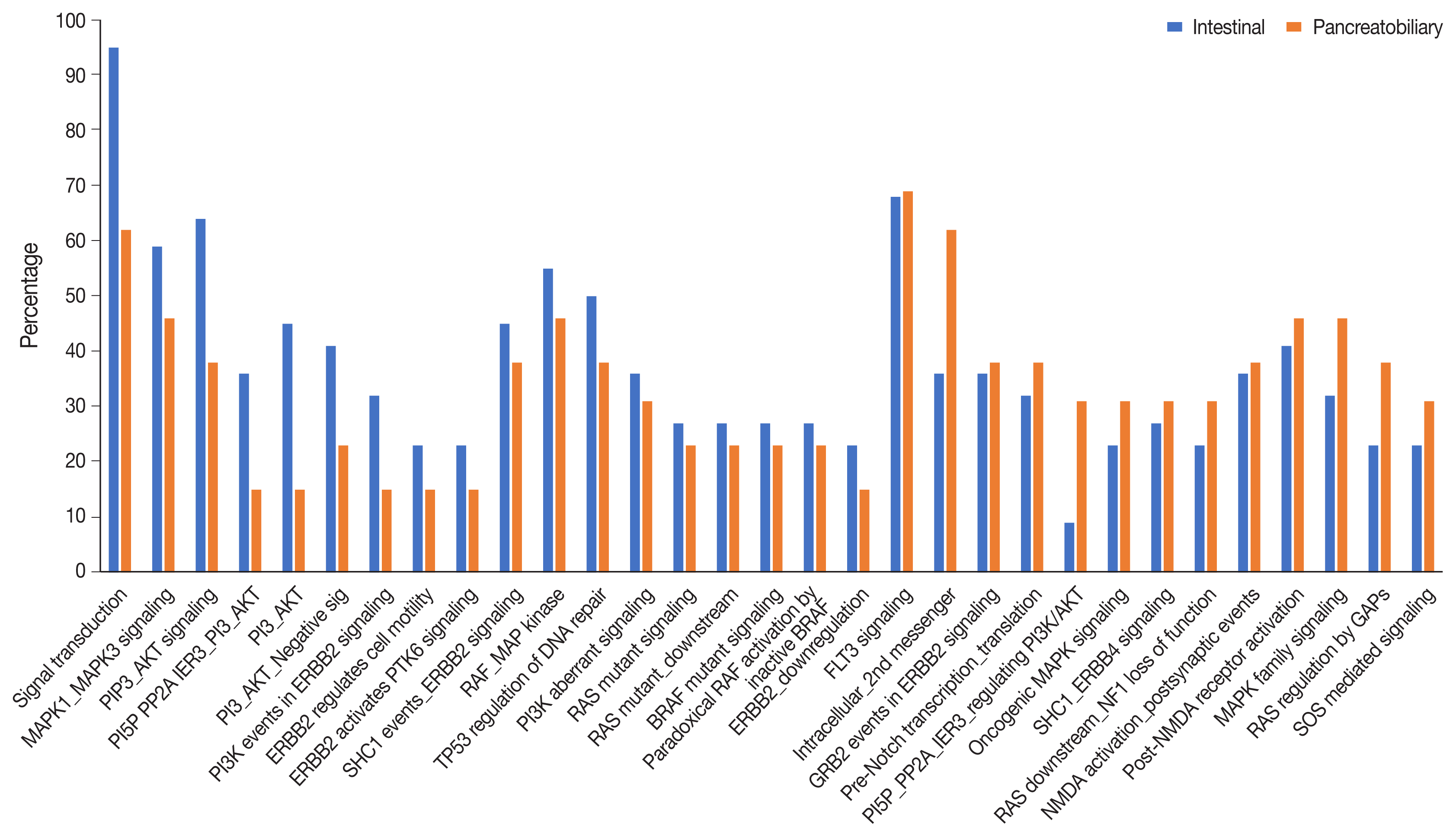
Fig. 8
Pathways involving the significantly most frequently mutated genes in intestinal and pancreatobiliary subtypes of ampullary carcinoma (p < .002).
![]()
Table 3
Major signaling pathways mutated in intestinal and pancreatobiliary differentiation of ampullary cancer
![]()
Tumor mutation burden (TMB) compared between the two differentiation types showed that INT type had 1.6 mutations/ Mb, while PB type had 0.8 mutations/Mb, with both of them falling into the low TMB group (p = .294).
DISCUSSION
The morphology of AC has emerged as an important prognostic factor in recent years and its subtyping as INT and PB types based on differentiation has shown INT type to have a better prognosis than PB type (p = .01) [10]. High-throughput sequencing techniques have revolutionized the genetic landscape of cancers. We studied the genetic map of AC and evaluated the pattern of mutational status according to morphologic subtype through WES. Major somatic mutations were found in nine genes in >10% of cases, similar to Lundgren et al. [13], who found TP53 and KRAS to be the most commonly mutated genes in their cohort. Genes harboring somatic mutations in > 10% patients of AC in the present study were TP53 (32.4%), KRAS (27.0%), KMT2 complex (24.3%), SMAD4 (21.6%), APC (18.9%), ErbB complex (16.2%), EphA complex (13.5%), Notch complex (13.5%), CTNNB1 (10.8%), and FAT1 (10.8%) genes. Other potentially targetable genes of ROS1, ARID2, and TAP1 were observed but at a lower frequency. The mutation profile of AC in the present study exhibited a similar spectrum to that reported in the two WES studies by Yachida et al. [11] and Gingras et al. [12], as well as studies with targeted panels of genes by Lundgren et al. [13] and Perkins et al. [14] with minor variations. Unlike Perkins et al. [14] who found mutations in CDKN2A and FBXW7 in 4% of cases and Lundgren et al. [13] who discovered CDKN2A mutations, we found FBXW7 in 2% of cases and CDKN2A mutations in none of the cases. CDKN2A mutations also were reported by Yachida et al. [11] and Gingras et al. [12], although at a lower frequency (4%), which was not reflected in our study population. One of the major findings reported by both these authors was mutation of the ELF3 driver gene (12%–13.3%), which was not detected in our study. In another study, Overman et al. [15] studied gene expression with a 92-gene classifier and found IRX3, PYCR1, and TMPRSS3 to be significantly associated with overall survival and relapse-free survival. However, pathogenic mutations in these genes were not found in the present study. Although there is a significant overlap between the genetic profiles of the present study and those of previous studies, there are certain differences as well, which could be attributed to the type of cancers studied (ampullary vs. periampullary), number of cases included (37 to 175), type of profiling methodology (whole exome vs. targeted panel comprising of a variable number of genes for somatic mutation vs. gene expression), and the ethnic diversity of the population studied.
Even though INT differentiation carries a better prognosis, it represents a genetically more unstable phenotype than PB differentiation (78 vs. 51 mutated genes), with a higher mutation frequency in APC, SMAD4, CTNNB1, TP53, EphA, KMT2, and Notch complex of genes, while ROS1 and TAP1 mutations were more frequent in PB differentiation. The KRAS and ErbB complex of genes harbored mutations in nearly equal frequency in both INT and PB differentiation. Lundgren et al. reported APC and ERBB3 mutations exclusively in the INT type of AC, which was reflected in our study with two cases of ERBB3 mutations exclusively found in INT differentiation. However, APC mutations were observed in both but were more frequent (27.3% vs. 7.8%) in the INT type of AC [13]. The genetic profiles of AC in different studies are presented in Table 4.
Table 4
Comparison of genetic profile studies in ampullary cancer
| Characteristic | Yachida et al. [11] | Gingras et al. [12] | Perkins et al. [14] | Lundgren et al. [13] | Present study |
|---|---|---|---|---|---|
| No. of cases studied | 172 (ampullary Ca) | 160 (ampullary 98, distal CBD 44, duodenal 18) | 91 (ampullary Ca) | 175 (ampullary 70, distal CBD 45, HOP 46, duodenal 14) | 37 (ampullary Ca) |
| Methodology | WES in 60 cases with mean coverage 188 ×, targeted 92-gene panel sequencing in 113 cases | WES with mean coverage 120 × | Targeted 50-gene panel | Targeted 70-gene panel | WES with mean coverage 200 × |
| No. of genes significantly mutated in > 10% patients | 24 | 19 | 18 | 9 | 18 |
| Gene (%) | |||||
| TP53 | 55.8 | 58 | 38 | 50 | 32.4 |
| KRAS | 47.6 | 55 | 46 | 46.1 | 27 |
| KMT2 family | - | - | - | - | 24.3 |
| SMAD4 | 16.3 | 20 | 9 | 14 | 21.6 |
| APC | 33.7 | 27 | 15 | 10 | 18.9 |
| ErbB family | 22 | 6 | 2 | 13 | 16.2 |
| EphA family | 18.6 | - | - | - | 13.5 |
| Notch family | - | - | - | - | 13.5 |
| CTNNB1 | 23.2 | 13 | - | - | 10.8 |
| ARID2 | 15.7 | 12 | - | - | 8.1 |
| PIK3CA | 5.8 | 18 | 14 | - | 5.4 |
| CDKN2A | 4 | 4 | 4 | 11.5 | 0 |
| FBXW7 | 5.8 | 6 | 4 | - | 2 |
| ELF3 | 13.3 | 12 | - | - | 0 |
| Major signaling pathway (%) | |||||
| TP53 | 58 | 68 | - | - | 38 |
| RAS-MAPK | 65.7 | 70 | - | - | 57 |
| PI3K-AKT | 11.6 | - | - | 49 | |
| WNT | 55.8 | 49 | - | - | 32 |
| TGF-β | 28.5 | 40 | - | - | 24 |
| Chromatin remodeling | 25.6 | 44 | - | - | 16 |
![]()
The major signaling pathways were grouped into six broad categories similar to those of Yachida et al. [11] and Gingras et al. [12]. The combined RAS-RAF-MAPK/PI3-AKT pathway was the major signaling pathway (70%) altered in AC, followed by TP53 (~38%), WNT signaling (~32%), TGF-β (24%), and the chromatin remodeling complex pathways (16%). The INT differentiation harbored mutations predominantly in the combined RAS-RAF-MAPK/PI3-AKT pathway in ~82% of cases, with PI3-AKT mutations more common (63.6%) than RAS-RAF-MAPK mutations (59.1%). The next most common pathways to be mutated in INT type were WNT (45.4%), TP53 (41.0%), TGF-β (22.7%), and the chromatin remodeling complex pathways (13.6%). The PB differentiation also harbored mutations predominantly in RAS-RAF-MAPK/PI3-AKT (46.2%) but at a significantly lower frequency (p = .02), and RAS-RAF-MAPK mutations were more frequent (46.2%) than PI3-AKT mutations (30.7%). The mutation frequencies of the TP53 pathway (38.4%) and the TGF-β pathway (22.7%) were similar in PB differentiation to that of INT type, while the WNT pathway (15.4%) had a much lower frequency, with the chromatin remodeling complex pathway (23.1%) having a higher mutation frequency in PB differentiation.
Yachida et al. [11] (WNT, TGF-β, PI3K, RTK-RAS, and p53-Rb signaling) and Gingras et al. [12] (TP53/cell division, RAS/PI3K, WNT, TGF-β, and chromatin remodeling pathways) in their WES studies observed a similar spectrum of mutated pathways to the present study. The WNT signaling pathway was reported as most frequently mutated in INT differentiation (67% in Gingras et al. [12], 76% in Yachida et al. [11]). However, it was the third most common pathway to be mutated in the INT type in the present study, after PI3-AKT and RAS-RAF-MAPK signaling. The oncogene panel (CHPv2 – Ampliseq, ABI) of Perkins et al. [14] found WNT to be the most commonly altered pathway in INT type, with mutations of KRAS and TP53 signaling to be more frequent in the PB type, similar to Yachida et al. [11,14]. TP53 mutations in our study had similar mutation frequency in the two types of differentiation, in contrast to those in the literature, where it has been reported to be more frequent in PB differentiation [11–14]. The Cancer Genome Atlas (TCGA) database states similarities between the molecular profile of INT differentiation and colorectal cancer harboring frequent APC mutations and WNT pathway alterations and that of PB differentiation and pancreatic adenocarcinoma harboring frequent TP53 and KRAS mutations [16,17].
The genetic profile of AC and those reported in the literature using WES or an oncogene panel through high-throughput sequencing suggests distinct molecular trends of histological INT and PB subtypes, although with significant overlap. These molecular trends might be important for guiding the therapeutic decision-making process. A better understanding of the molecular profile and the emergence of newer targeted agents could lead to better treatment outcomes in AC, as seen in other cancers. Future studies on the relationship of the genetic profile and response to adjuvant therapeutic regimens will serve to improve and devise more precise therapeutic regimens for AC patients, which are currently extrapolated from the established treatment protocols for pancreatic, biliary, and intestinal cancers.
To summarize, we present the first WES data in AC from India providing a genetic map of INT and PB subtypes. Approximately 86% of AC patients harbor one or more targetable mutations. The WES data suggest that INT type cancers are more unstable genetically than PB types, and that both involve mutations in tumor suppressor, transcription factor, and chromatin remodeling complex genes. The most frequent targetable mutations were found in KRAS, CTNNB1, TP53, APC, and SMAD4 genes. Our data suggest the PI3/AKT and RAS/MAPK kinase pathways to be predominantly mutated in AC, suggesting support of primary targeting of the PI3/AKT pathway in INT and the RAS/MAPK kinase pathway in PB type AC. Mutational profiling would enable better patient stratification and help to identify potential responders to targeted or personalized therapies in these cancers.
 XML Download
XML Download