

 PDF
PDF Citation
Citation Print
Print



Serrated lesions and polyps of the colorectum include all nonmalignant epithelial neoplastic lesions showing serrated morphology in the crypt epithelium. Until recently, serrated colorectal lesions were largely classified into three categories: hyperplastic polyp (HP), sessile serrated adenoma/polyp (SSA/P), and traditional serrated adenoma (TSA). However, since the publication of the previous 2010 World Health Organization (WHO) classification, many studies have improved our knowledge of serrated colorectal lesion pathology. In the recently updated 2019 WHO classification, there have been important changes in classification, terminology, and diagnostic criteria for serrated colorectal lesions. In this review, we briefly summarize three major components of the pathology of serrated lesions: (1) updates on the 2019 WHO classification of serrated lesions, (2) updates on morphologic variants and dysplasia of serrated lesions, and (3) the molecular pathology of serrated lesions.
UPDATES IN THE 2019 WHO CLASSIFICATION OF SERRATED COLORECTAL LESIONS
Classification, terminology, and diagnostic criteria for serrated lesions/polyps of the colorectum are being revised, and their clinical implications and molecular features have also been newly discovered or modified. The WHO classification of tumors of the digestive system was recently updated to the 5th edition [1]. Compared to the previous edition, the 5th edition has demonstrated several notable changes in the section on serrated colorectal lesions/polyps.
Changes in the terminology and categorization of serrated colorectal lesions
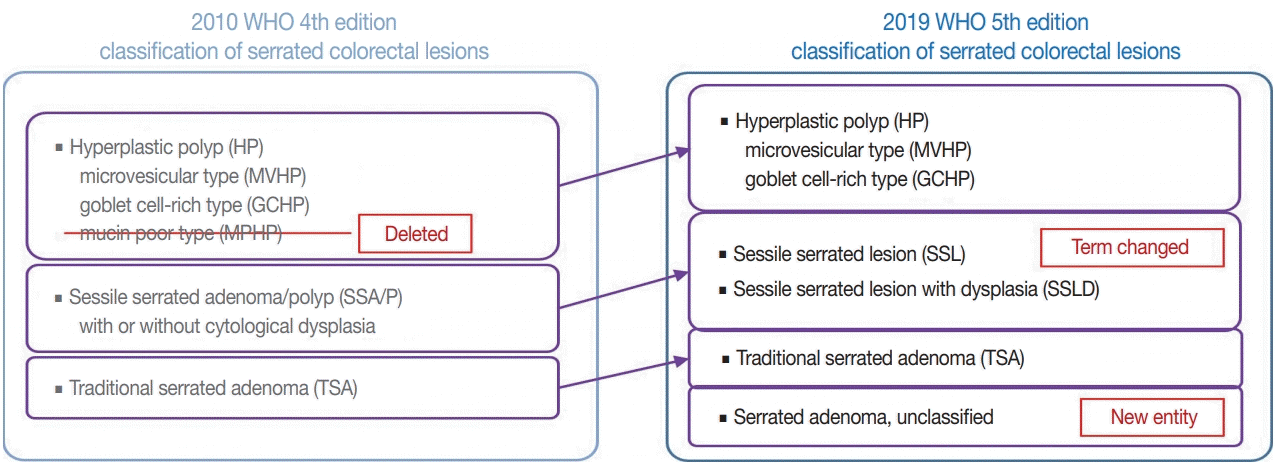
There are major and minor changes in the terminology and categorization of serrated colorectal lesions. Alterations in the terminology and categorization of serrated colorectal lesions in the new WHO classification (5th edition) are summarized in Fig. 1. The most important change in terminology is the adoption of the new diagnostic term “sessile serrated lesion” (SSL), which refers to serrated premalignant lesions that were formerly called “sessile serrated adenomas” (SSAs) or “sessile serrated polyps” (SSPs) (Fig. 1) [1]. The rationale for replacing the term “adenoma” with “lesion” is based on the fact that a considerable number of SSLs do not show morphologic dysplasia, an essential component of classical “adenoma” in the colorectum [2]. Moreover, because some SSLs may not show a polypoid appearance, the term “polyp” is unsuitable [1]. There was also a minor change in the SSL category. Although SSLs (SSA/Ps) with cytologic dysplasia have already been described as an advanced subtype of SSL in the previous edition, the new WHO classification more clearly categorizes “SSL with dysplasia” (SSLD) as an established diagnostic terminology (Fig. 1) [1]. Another change in the new classification is the elimination of mucin-poor HP (MPHP) among the HP subtypes (Fig. 1) [1]. According to the previous WHO classification, HPs can be classified into three subtypes: microvesicular HP (MVHP), goblet cell-rich HP (GCHP), and a minor subtype, MPHP [3]. However, in the new WHO classification, only MVHP and GCHP remain among the HP subtypes. In fact, subtyping of HPs into MVHP or GCHP is practically unnecessary in pathologic diagnosis because the clinical significance of HP subtyping has not been proven [4]. The last change in terminology in the new WHO classification of serrated lesions introduces the new diagnostic entity “unclassified serrated adenoma” (or “serrated adenoma, unclassified”) (Fig. 1) [1]. In fact, pathologists have occasionally encountered problematic colorectal polyp cases showing both dysplasia and serrated architecture, but these cannot be clearly classified as SSL, TSA, or conventional adenoma. Thus, these ambiguous adenomas with serrated morphology, including the recently suggested serrated tubulovillous adenoma (sTVA) category (described below), can be diagnosed as unclassified serrated adenoma (USA) according to the new WHO classification. Terminology and categorization of TSAs were maintained without revision from the 4th to the 5th edition of the WHO classification (Fig. 1).
Changes in the diagnostic criteria for SSLs
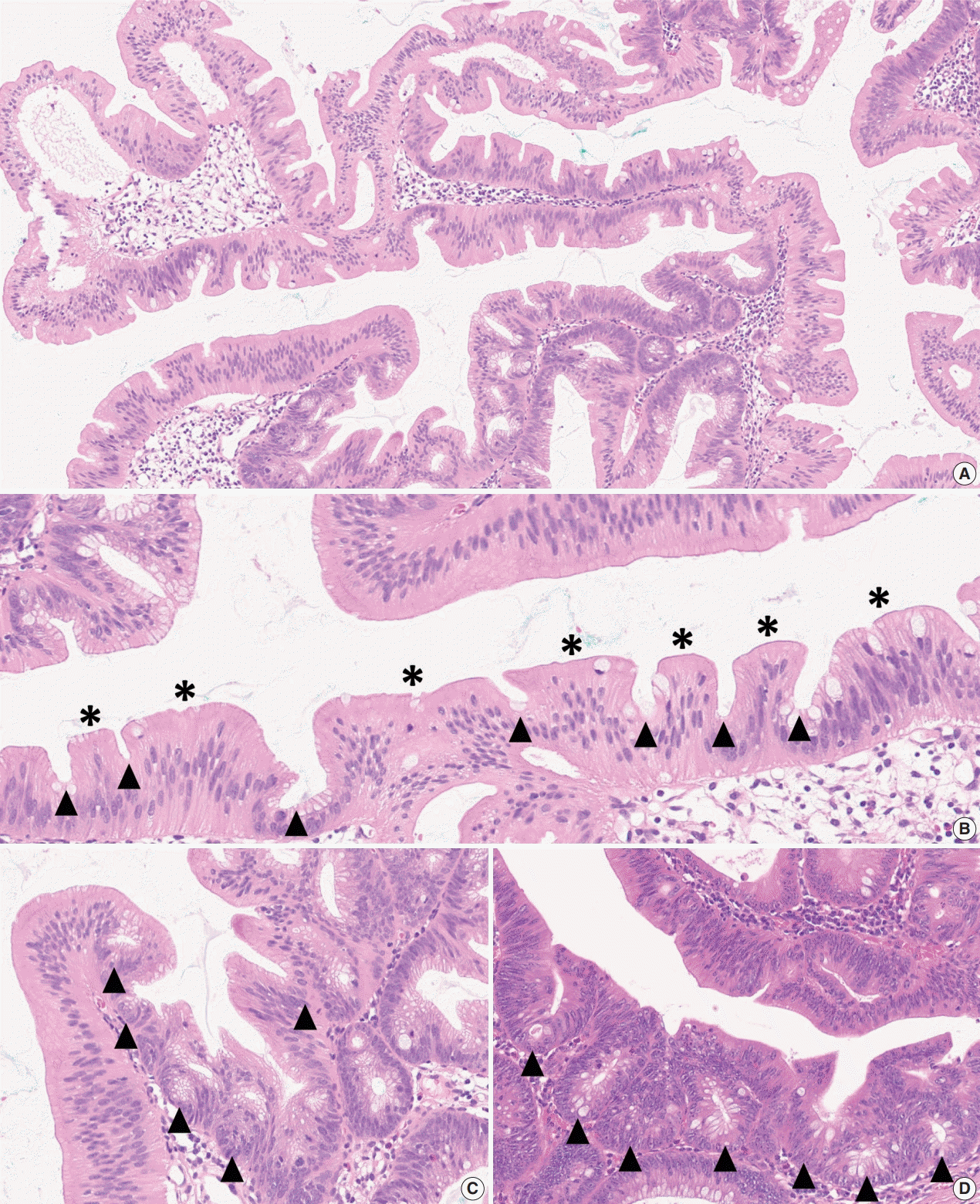
The diagnostic criteria for SSLs in the 5th WHO classification have become clearer than those of the previous edition. According to the 4th WHO classification, two or three typically distorted serrated crypts in a polyp might be necessary to diagnose SSL [3]. Because “two or three” was somewhat unclear for a cut-off value to establish definitive diagnosis, this suggestion occasionally induced confusion, especially when there were two distorted serrated crypts in a non-dysplastic serrated lesion. The criteria were occasionally interpreted as two contiguous or three dispersed typical crypts. After the publication of the 4th WHO classification in 2010, an American expert panel suggested that only one distorted serrated crypt might be sufficient for the diagnosis of SSL [5]. The updated 2019 WHO classification described this recommendation as the minimum requirement for SSL diagnosis [1]. Detailed diagnostic criteria for SSL based on the new WHO classification are summarized in Table 1. In detail, a serrated colorectal lesion must show an overall distorted crypt architecture to be diagnosed as an SSL. The architecturally distorted serrated crypt pathognomonic for SSL should satisfy one or more of the following morphologic features: (1) horizontally growing crypt along the muscularis mucosa, (2) dilated crypt base, (3) serrated morphology throughout the crypt including the base, and (4) asymmetrical proliferative features of the crypt (Table 1) [1]. The 5th WHO classification exclusively introduced the presence of “one or more” unequivocal distorted serrated crypts as a diagnostic criterion for SSLs [1], which was suggested by an American expert panel [5], instead of the “two or three” criteria of the 4th WHO classification [3]. The authors of the new WHO classification also emphasized that a distorted serrated crypt should be morphologically “unequivocal” for the diagnosis of SSL [1]. Serrated crypts with equivocally distorted architecture, including crypt branching and mild symmetrical dilatation of the crypt base, should not be included in the count of distorted serrated crypts for SSL diagnosis [1]. Examples of unequivocal and equivocal architecturally distorted serrated crypts in SSLs are shown in Fig. 2.
Changes in the diagnostic criteria for TSAs
As mentioned above, there is no remarkable change in the terminology or categorization of TSAs. However, there is a minor revision in the description of essential morphologic components for the diagnosis of TSAs. Since the 4th edition of the WHO classification, it has been understood that TSAs have three morphologic characteristics that distinguish them from other serrated lesions or conventional adenomas: (1) unique cytological features, including intense eosinophilic cytoplasm with pencillate nuclei, (2) unique serrated morphology, including slit-like serrations, and (3) a unique crypt architecture, including ectopic crypt foci (ECFs) (Fig. 3A–C). Among these, the cytological and serration features are still recognized as defining factors of TSAs (Fig. 3B). Previous references, including the 4th edition of the WHO classification, described that the presence of ECF could be pathognomonic for TSAs [3]. However, recent investigations have reported that ECFs can be found not only in TSAs but also in other adenomas, including tubulovillous adenomas (TVAs), villous adenomas (VAs), and sTVAs (Fig. 3C, D) [6,7]. Thus, ECF is currently not considered to be an exclusive feature of TSAs, and it is recommended that the diagnosis of TSAs based only on the presence of ECF should be avoided [1,4].
UPDATES ON MORPHOLOGIC VARIANTS AND DYSPLASTIC PATTERNS OF SERRATED LESIONS
In recent years, there have been several suggestions for novel morphologic variants of serrated colorectal lesions. Here, we review recently proposed morphologic variants including sTVA, mucin-rich variant of TSA (MrTSA), and superficially serrated adenoma (SuSA). These variants have not been widely accepted and are not officially included as diagnostic terms in the 2019 WHO classification. However, the molecular features and biological behaviors of these variants might be somewhat different from those of classic SSLs and TSAs. Therefore, to establish these variants as official subtypes of serrated lesions, clinicopathologic and molecular data of these variants should be further analyzed.
Serrated tubulovillous adenoma (sTVA)
sTVA was first defined by Bettington et al. [7], who stated that sTVA can be diagnosed when a polyp meets all the following criteria: (1) villous component in more than 25% of the polyp, (2) serrated morphology in more than 50% of the polyp, and (3) TSA-type cytological features and slit-like serrations in less than 10% of the polyp. Bettington et al. [7] found that sTVAs were larger and more likely to be proximal; they were also molecularly associated with CpG island methylation and KRAS mutations more than conventional TVAs. In addition, sTVAs were more likely to be proximal and were associated with less CpG island methylation and more frequent β-catenin nuclear expression than TSAs [7]. There were no cases showing MLH1 loss or BRAF mutation among the studied sTVAs [7]. These findings collectively indicate that sTVAs may be precursors of KRAS-mutated, microsatellite-stable (MSS) colorectal carcinomas (CRCs).
Mucin-rich TSA (MrTSA)
MrTSA was first described by Kalimuthu et al. [8]. MrTSA can be defined as a TSA showing ≥50% goblet cells or mucinrich cells with a goblet cell/eosinophilic absorptive cell ratio of at least 1:1 [8]. Compared to classic TSAs, MrTSAs are characterized by variable growth patterns, a lower frequency of ECFs, and more intraepithelial lymphocytes [8]. Furthermore, the molecular characteristics of MrTSAs were also analyzed by Hiromoto et al. [9], who compared KRAS/BRAF mutation profiles and immunohistochemical expression statuses of MrTSAs to those of classic TSAs and sTVAs. In this study, MrTSAs demonstrated retained MLH1 expression, frequent BRAF mutations, and rare KRAS mutations [9]. These findings suggest that the majority of MrTSAs may be precursors of BRAF-mutated MSS CRCs, an aggressive molecular subtype of CRC.
Superficially serrated adenoma (SuSA)
SuSA was most recently suggested by Hashimoto et al. [10] as a polyp showing characteristic mixed adenomatous and serrated features, including straight adenomatous glands with serrations confined to the superficial portion. Molecularly, SuSAs display high frequencies of KRAS mutations and RSPO fusions [10]. Because concurrent KRAS mutations and RSPO fusions are distinct molecular features of a subset of TSAs [11], it is thought that SuSAs may be biologically connected to KRAS-mutated TSAs and, like sTVAs, may also be precursors of KRAS-mutated MSS CRC. Consistent with these findings, a case report recently described that a sigmoid colon carcinoma derived from SuSA molecularly demonstrated a KRAS mutation and a RSPO2 fusion [12].
Unclassified serrated adenoma (USA)
Based on the updated 2019 WHO classification, both sTVAs and SuSAs may be included in the newly defined USA category. As mentioned above, the new WHO classification proposed that the USA can be defined as an unclassifiable dysplastic polyp with serrated architecture [1]. However, some of the newly defined serrated lesion variants including sTVAs and SuSAs demonstrate clinicopathologic and molecular features different from those of other classic serrated lesions; they may also be associated with transitional or mixed molecular profiles between serrated lesions and conventional adenomas. Although both sTVAs and SuSAs may be common precursors of KRAS-mutated MSS CRCs, there are also differences in detailed morphologic and molecular characteristics between sTVAs and SuSAs. These findings indicate that the USA category may need to include fairly heterogeneous serrated lesions. Therefore, to avoid using USA as a wastebasket diagnosis, it is expected that the USA category will be further subclassified into sTVA, SuSA, and other new variants based on their morphologic and molecular differences.
Pathologic issues in morphologic dysplasia of serrated lesions
Although SSLs have been regarded as precursors to CRC, only a small proportion of overall SSLs show morphologic dysplasia (SSLDs). Moreover, some dysplasia patterns found in SSLDs differ considerably from the typical dysplastic features of colorectal conventional adenomas. It has been generally recognized that there are two distinct dysplasia subtypes in SSLDs: intestinal (adenomatous) and serrated [1,3,13]. Intestinal dysplasia in dysplastic serrated lesions is morphologically similar to the low-grade dysplasia found in conventional adenomas. Intestinal dysplasia in SSLDs is mainly characterized by typical cytological features including elongated, pseudostratified, hyperchromatic nuclei, and basophilic cytoplasm [1,3,13]. In contrast, serrated dysplasia in dysplastic serrated lesions is morphologically characterized by cuboidal cells, eosinophilic cytoplasm, increased mitoses, and nuclear atypia with vesicular nuclei and prominent nucleoli [1,3,13]. In both intestinal and serrated dysplasia, architectural complexity has not been considered as an essential factor for diagnosis, indicating that SSLDs can be diagnosed only based on cytological dysplasia within any portion of the crypt epithelium of SSLs. Of course, SSLDs can accompany various levels of architectural abnormalities; however, these architectural features generally do not change the simple diagnostic term “SSLD” because subtyping or grading of SSLDs has not yet been officially recommended.
In addition to the traditional dichotomous subtyping of dysplastic patterns in SSLDs, Liu et al. [14] proposed a novel classification of four different dysplastic patterns in SSLDs: (1) minimal deviation, (2) serrated, (3) adenomatous, and (4) not otherwise specified [14]. Minimal deviation dysplasia demonstrates only minor cytological and architectural changes but is mostly accompanied by loss of MLH1 expression (91%) [14]. Serrated dysplasia is architecturally characterized by tightly packed small glands with decreased serrations, and cytologically demonstrates frequent mitoses, atypical vesicular nuclei, and prominent nucleoli. Adenomatous dysplasia is similar to the dysplasia morphology of conventional adenomas. Liu et al. [14] reported that loss of MLH1 expression was rare in both serrated and adenomatous dysplasia (13% and 5%, respectively). Dysplasia not otherwise specified includes all dysplastic patterns not fulfilling the criteria of the above three dysplasia patterns and is the most common dysplasia subtype in SSLDs (79%) [14]. In contrast to serrated and adenomatous dysplasia, loss of MLH1 expression was frequently found in dysplasia not otherwise specified (83%) [14]. These four dysplasia subtypes have not yet been officially adopted by the WHO classification or other guidelines, and currently, there is little need to classify the four dysplasia patterns in the practical diagnosis of SSLDs. In future studies, the reproducibility of this dysplasia classification system should be further evaluated, and differential clinicopathologic and molecular implications of the four dysplasia patterns should be further elucidated.
As briefly mentioned above, grading of dysplasia (low-grade vs. high-grade), an essential component for the diagnostic description of conventional adenomas, is not recommended for dysplastic serrated lesions including SSLD, TSA, and USA [1]. This is because there are various issues related to morphologic heterogeneity, low reproducibility, and uncertain clinical implications. However, we believe that if a pathologist observes definite high-grade dysplastic (HGD) features or intramucosal carcinoma (IMC) components in a serrated lesion, based on the morphologic criteria applied in conventional adenomas, then this should be described in the diagnostic report. The new WHO classification also recommends that the HGD component should be reported separately when it is found in a TSA [1]. There is no consensus regarding the grading of dysplasia in serrated lesions. However, determination of HGD based on the traditional criteria for conventional adenomas may also be acceptable in serrated lesions because both morphologic characteristics and the risk of further invasive progression of advanced lesions may not differ significantly between the two. Conventional adenomas with HGD are characterized by combined cytological atypia (loss of polarity, marked enlargement of nuclei, prominent nucleoli, and occasional atypical mitoses) and architectural complexity (crowded, cribiforming, and irregularly branching glands with or without intraluminal necrosis) [1]. To determine HGD in conventional colorectal adenomas, architectural abnormalities are generally considered to be more critical than cytological features. Accompanying IMC can be diagnosed when there are atypical glands invading the lamina propria without invasion beyond the muscularis mucosa in an adenoma [1,15]. To establish surveillance and treatment strategies suitable for advanced serrated colorectal lesions, detailed differences in the morphologic, molecular, and prognostic features of HGD/IMC components between serrated lesions and conventional adenomas should be further investigated.
UPDATES ON THE MOLECULAR PATHOLOGY OF SERRATED LESIONS
Molecular pathogenesis of SSLs
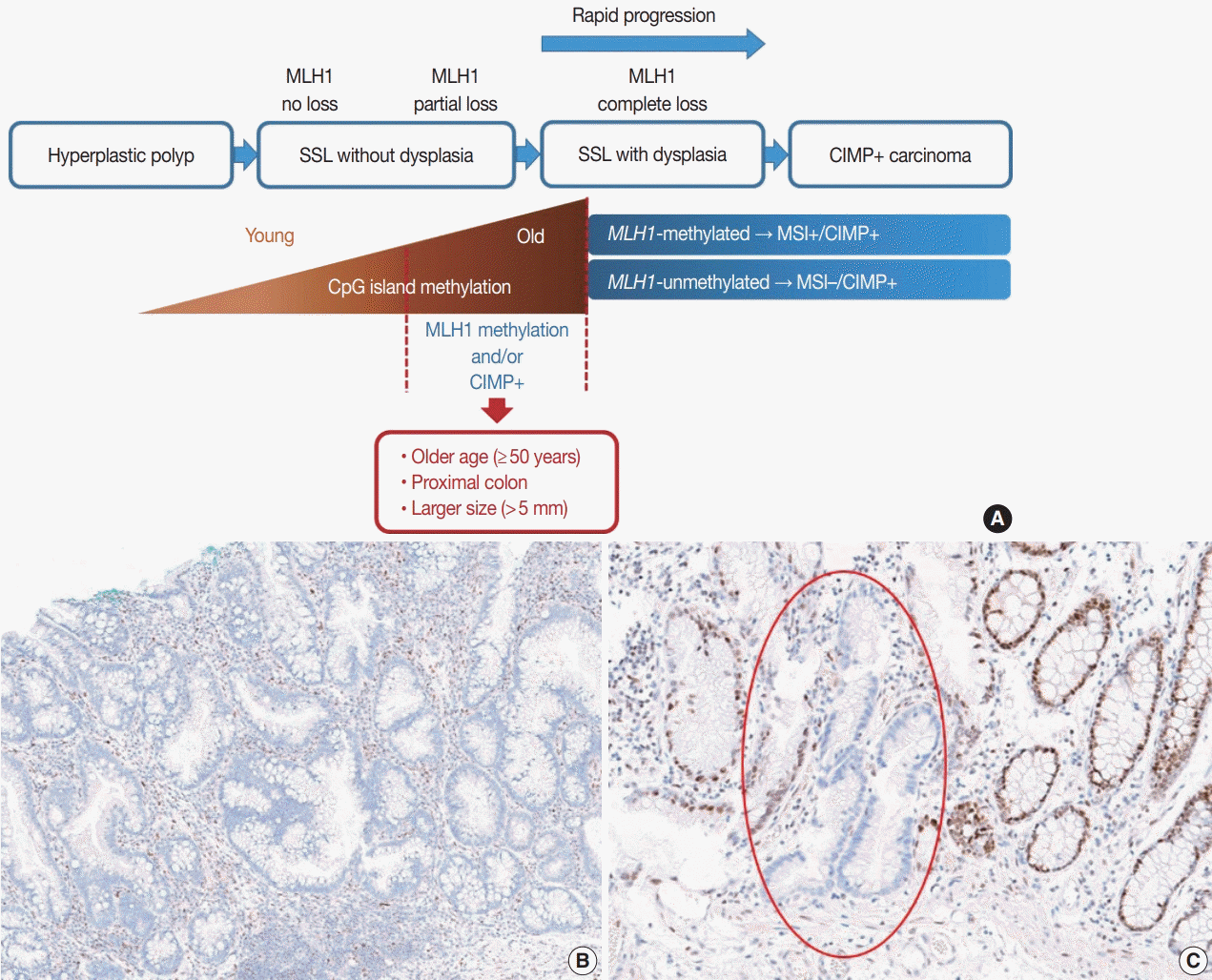
It is hypothesized that most CRCs which develop through SSLs molecularly display CpG island methylator phenotypepositivity (CIMP+ or CIMP-high), which is characterized by silencing of many cancer-related genes by extensive promoter CpG island hypermethylation [16,17]. The MLH1 gene is a DNA mismatch repair (MMR) gene and is also frequently methylated under the CIMP+ condition [18]. MLH1 silencing by promoter methylation causes high microsatellite instability (MSI-high), indicating an MSI-positive (MSI+) status that is characterized by genome-wide microsatellite sequence alterations and many consequential mutations [18]. CIMP+ with or without MLH1 methylation is regarded as a major molecular hallmark of the transformation of SSLs to more advanced lesions (SSLDs or carcinomas) (Fig. 4A). If MSI-high is induced in an SSL by MLH1 silencing, then this lesion will almost inevitably progress into an SSLD, which can rapidly transform into a carcinoma due to the high oncogenic pressure of high mutational burden (Fig. 4A). An interesting case report regarding the rapid malignant transformation of an SSL was recently published [19]. The rapid progression of SSLDs may explain why the detection rate of SSLDs among biopsied or resected colorectal polyps is generally very low; this may be one cause of interval cancers in the proximal colon [20].
Recent investigations including our previous study suggested that both MLH1 methylation and CIMP+ occur almost exclusively in proximal, large SSLs in elderly patients (Fig. 4A) [21-23]. Liu et al. [22] examined 448 SSLs and found that the proportion of CIMP+ abruptly increased in SSL patients 51 or older compared to younger patients. According to the study, CIMP+ SSLs were significantly associated with older age and proximal colonic location, but were not correlated with patient sex or lesion size [22]. In our study, we tested 132 non-dysplastic SSLs and defined SSLs with CIMP+ and/or MLH1 methylation as molecularly high-risk lesions. These high-risk SSLs were found exclusively in the older age group (≥50 years; 100%) and in the proximal colon (100%), which was statistically significant [21]. Although our findings were similar to the results of Liu et al. [22], we also found that histologically measured lesion size, but not endoscopically measured size, was significantly associated with CIMP+/MLH1 methylation in non-dysplastic SSLs; the high-risk SSLs were found exclusively in SSLs which were >5 mm on histology [21]. Therefore, we concluded that SSLs with CIMP+ and/or MLH1 methylation are concentrated in a specific subgroup of SSLs satisfying all the following conditions: older age (≥50 years), proximal colonic location (cecum, ascending colon, or transverse colon), and histologically larger polyp size (>5 mm) (Fig. 4A).
Most SSLDs harboring MLH1 promoter methylation typically show complete loss of MLH1 immunohistochemical expression in their dysplastic crypts (Fig. 4B) [21,24]. Many nondysplastic SSLs are MLH1-unmethylated and display retained expression of the MLH1 protein. However, we recently found that partial loss of MLH1 expression (involving a single or a few non-dysplastic crypts) can occasionally be observed in non-dysplastic SSLs (Fig. 4C) [21]. These unusual non-dysplastic SSLs with partial MLH1 loss demonstrated MLH1 promoter methylation; however, they had lower levels of MLH1 methylation than MLH1-methylated SSLDs with complete loss of MLH1 expression (Fig. 4B, C) [21]. This novel finding was supported by another recent study, which reported that 71 out of 400 (18%) cases demonstrated loss of MLH1 expression in their non-dysplastic crypts [25]. Collectively, partial loss of MLH1 expression in non-dysplastic SSLs can be a sign of impending dysplastic change and may be a biomarker to screen for molecularly advanced lesions among non-dysplastic SSLs.
The BRAF V600E mutation, along with CIMP+, is regarded as a molecular hallmark in the colorectal sessile serrated neoplasia pathway [26-28]. It is known that CIMP+ is tightly associated with the BRAF mutation in CRCs [29], indicating that both CIMP+ and the BRAF mutation originate from common premalignant lesions (SSLs) in the colon and rectum. Although it has been strongly suspected that the BRAF mutation and CIMP+ might synergistically impact carcinogenesis, the detailed mechanism underlying their interaction is unclear. Fang et al. [30,31] previously suggested that the BRAF oncoprotein might promote CpG island methylation in multiple gene promoters in CRC cells through increased promoter binding of MAFG, a transcriptional repressor. However, this finding has not yet been validated by other studies. Recently, Tao et al. [32] reported important clues for the relationship between the BRAF mutation and CIMP+ in colorectal carcinogenesis. In their experimental study, agingrelated hypermethylation induced sensitivity of mouse colon organoids to BRAF mutation-induced oncogenic transformation [32]. This suggests that a BRAF mutation is not a prerequisite for CIMP development in SSLs. The finding is also consistent with the real-world observations that CIMP+ SSLs and subsequent CIMP+/BRAF-mutated CRCs occur almost exclusively in older patients.
According to data from Western countries including the United States, Canada, Australia, Germany, Austria, and Switzerland, frequencies of BRAF V600E mutations in SSLs range from 63% to 100% (Supplementary Table S1) [22,24,33-41]. However, in East Asian countries, including South Korea, Japan, and China, BRAF mutation frequencies in SSLs have been reported to be relatively lower, ranging from 14% to 86% (Supplementary Table S1) [42-55]. Using these data, we conducted a pooled analysis to directly compare the frequencies of BRAF mutations in SSLs between Western and Eastern countries (Table 2). Overall, BRAF mutations were found in 91% (932 of 1,028) of SSLs from Western countries and 76% (798 of 1,048) of SSLs in Eastern countries (Table 2). Interestingly, KRAS mutations, known to be mutually exclusive with BRAF mutations in tumors, were more frequently found in SSLs from Eastern countries (6%, 65 of 1,053 SSLs) compared to Western countries (2%, 22 of 988 SSLs) (Table 2). These regional differences in BRAF and KRAS mutation frequencies in SSLs were all statistically significant (both p<.001) (Table 2). Although it is generally accepted that nearly all Western colorectal SSLs have BRAF mutations, there are likely ethnic differences in BRAF and KRAS mutation profiles in SSLs, based on the results of our pooled analysis. Unsurprisingly, the ethnic differences in BRAF mutation frequency in SSLs can be linked to a similar tendency in the molecular profiles of CRCs. In fact, we previously reported that CRCs from East Asian patients have lower BRAF mutation frequencies than those from Western populations [18]. In brief, it is reasonable to suggest that the lower frequency of BRAF mutations in SSLs from East Asian patients is directly reflected in the lower frequency of BRAF mutations in CRCs of the same regional population because SSLs are major precursors of BRAF-mutant CRCs (Fig. 5A).
Several previous studies analyzed RNA and/or protein expression profiles of SSLs. Caruso et al. [36] first found upregulation of cathepsin E (CTSE) and trefoil factor 1 in SSLs. It is also known that gastric-type mucins, including mucin 6 (MUC6) and mucin 5AC (MUC5AC), can be aberrantly expressed in SSLs and HPs [56-58]. Gonzalo et al. [59] suggested that annexin A10 (ANXA10) could be a potential marker of SSLs. Various other proteins normally expressed in the gastric mucosa such as v-set and immunoglobulin domain containing 1 (VSIG1) and trefoil factor 2 (TFF2) were frequently overexpressed in SSLs [60,61]. However, the practical utility of these gastric-type proteins in the diagnosis of SSLs has been limited because these markers can also be expressed in subsets of other serrated lesions, including TSAs and HPs [61]. Recently, Rickelt and colleagues published interesting data regarding a potential biomarker of SSLs [62]. In their study, agrin (AGRN) expression in the muscularis mucosa is a specific feature of SSLs, which contrasts with the absence of AGRN expression in the muscularis mucosa of other types of serrated lesions and conventional adenomas [62]. The utility of AGRN immunohistochemistry in the differential diagnosis between SSLs and other colorectal polyps should be further validated by independent studies using large-scale samples.
Molecular pathogenesis of TSAs
During the past decade, there has been remarkable progress in understanding the molecular features of TSAs. The molecular pathogenesis of TSAs can be largely divided into two pathways: the KRAS mutation pathway and the BRAF mutation pathway [63,64]. KRAS or BRAF mutations are found in more than 80% of TSAs [41,64], and whether KRAS or BRAF mutations occur in a TSA is mainly dependent on the location of the TSA. Most KRAS-mutated TSAs are found in the distal colon (descending and sigmoid colon) and rectum, whereas BRAF-mutated TSAs are more frequently located in the proximal colon (cecum and the ascending and transverse colon) [64]. Because TSAs are generally more prevalent in the distal rather than proximal colon [11], KRAS mutations are also more prevalent than BRAF mutations in TSAs. It has been suggested that BRAF-mutated TSAs may be derived from proximal colonic SSLs or HPs based on morphologic connectivity and molecular similarity. SSLs or HPs are often found as precursor lesions around TSAs, especially proximal TSAs, indicating that proximally-located, BRAF-mutated TSAs can be transformed from preexisting SSLs or HPs [64-67]. In terms of molecular features, BRAF-mutated TSAs of the proximal colon frequently show CIMP+, whereas KRAS-mutated TSAs of the distal colorectum generally display CIMP-low or CIMP-negative status [63,64]. Because CIMP+ is an epigenetic hallmark of advanced SSLs as described above, proximal TSAs commonly share two major molecular features with SSLs, namely, BRAF mutation and CIMP+, and these findings support the theory of transition from SSLs to TSAs in the proximal colon. Although MLH1 methylation is a frequent event in CIMP+ SSLDs, MLH1 methylation or complete loss of MLH1 expression is rarely found in proximal TSAs, even in advanced TSAs [64]; this suggests that most proximal, BRAF-mutated TSAs may be precursors of CIMP+/MSI− CRCs but not CIMP+/ MSI+ CRCs (Fig. 5A).
APC mutations are an initiating molecular hallmark and a main cause of Wnt signaling pathway activation in colorectal conventional adenomas; though these mutations are not frequent in TSAs, many TSAs show Wnt pathway activation, which can be confirmed by β-catenin expression in tumor cell nuclei [64,67,68]. Recent studies have identified that Wnt activation in TSAs may be caused more frequently by RSPO fusions or RNF43 mutations than by APC mutations [69]. Sekine et al. [69] previously reported that PTPRK-RSPO3 fusions and RNF43 mutations were found almost exclusively in TSAs compared to other conventional adenomas or serrated lesions. In their study, genetic alterations associated with Wnt activation including RSPO3 fusions, RNF43 mutations, APC mutations, and CTNNB1 mutations were mutually exclusive in TSAs [69]. Subsequent studies revealed that RSPO fusion-positive TSAs were significantly associated with distal location, larger size, and KRAS mutations, whereas RNF43 mutations were frequently found in BRAF-mutated TSAs [67,70,71]. Although PTPRK–RSPO3 fusions were a predominant form of RSPO fusions in TSAs, other minor fusions including NRIP1–RSPO2, EIF3E–RSPO2, and PIEZO1–RSPO2 fusions were also found in TSAs [70,72]. Interestingly, in TSAs with adjacent precursor lesions (SSL or HP), Wnt activation-related genetic alterations including RSPO fusions, RNF43 homozygous mutations, APC mutations, and CTNNB1 mutations were found only in the TSA component but not in the precursor component, indicating that Wnt activation may be critical for the development of TSAs from precursor lesions [67].
Like SSLs, TSAs express gastric-type proteins including ANXA10, VSIG1, CTSE, TFF2, and MUC5AC, although their positivity rates and the intensity or extent of expression are generally lower than those of SSLs or MVHPs [61]. Specific biomarkers for the differential diagnosis of TSAs have not been established. However, Sohier et al. [73] performed proteomic analysis using formalin-fixed, paraffin-embedded tissues of various types of serrated lesions and conventional adenomas and found that LEFTY1, an inhibitory protein of the Nodal/transforming growth factor β pathway, was overexpressed specifically in TSAs. Although this may be promising, more studies will be necessary to validate whether LEFTY1 immunohistochemistry is practically useful in the diagnosis of TSAs.
Serrated lesions as precursors of different molecular subtypes of CRCs
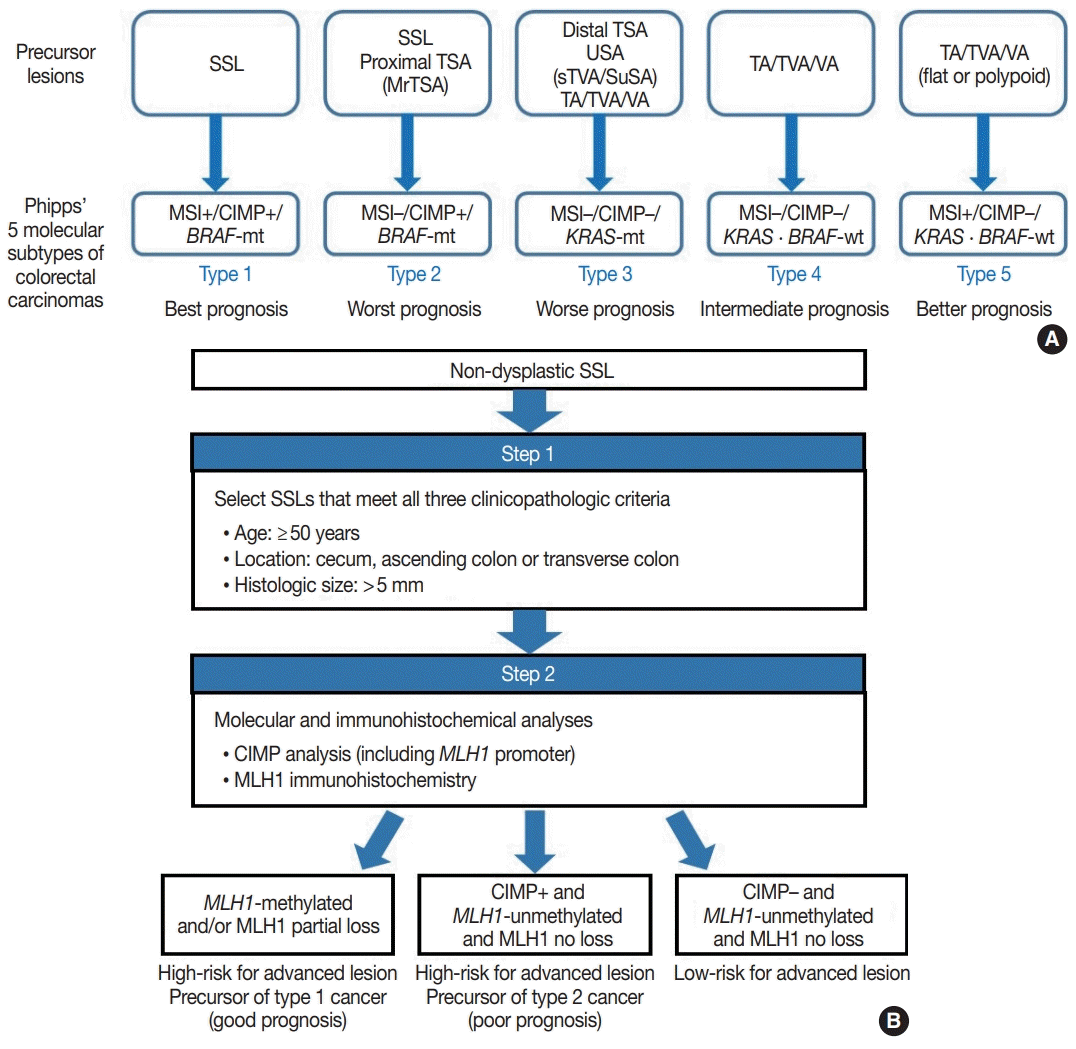
Because SSLs, TSAs, and their variants are precursor lesions of CRCs, and each shows unique molecular features, serrated premalignant lesions can be matched to their consequential molecular subtypes of CRCs. Phipps et al. [74,75] recently classified CRCs into five molecular subtypes based on combined MSI, CIMP, and KRAS/BRAF mutation profiles and successfully validated the prognostic significance of CRC molecular subtyping in large-scale cohorts. The five molecular subtypes were defined as follows: (1) type 1: MSI+, CIMP+, BRAF-mutated, KRAS-wildtype; (2) type 2: MSI−, CIMP+, BRAF-mutated, KRAS-wildtype; (3) type 3: MSI−, CIMP−, BRAF-wildtype, KRAS-mutated; (4) type 4: MSI−, CIMP−, BRAF-wildtype, KRAS-wildtype; and (5) type 5: MSI+, CIMP−, BRAF-wildtype, KRAS-wildtype (Fig. 5A) [74,75]. This molecular subtyping proved to be useful in the prognostication of CRCs. The prognosis of the five molecular subtypes was ranked (best to worst) as follows: type 1–type 5–type 4–type 3–type 2 (Fig. 5A) [74,75]. Type 1 CRCs indicate sporadic MSI+ (MSI-high) tumors caused by promoter methylation-associated MLH1 silencing, whereas the majority of type 5 CRCs are hereditary MSI+ tumors that arise in the setting of Lynch syndrome, which is genetically defined by germline mutations in one of the MMR genes. Because both type 1 and type 5 CRCs are molecularly MSI-high, which is well-established as a favorable prognostic factor in CRC [18], patients with these tumors generally show good survival. Sporadic MLH1 methylation with CIMP+ and BRAF mutation occurs almost exclusively in SSLs among colorectal premalignant lesions. Thus, SSLs can be considered as unequivocal precursors of type 1 CRCs (Fig. 5A). Most precursor lesions in Lynch syndrome-associated CRCs are histologically conventional-type adenomas with grossly flat or polypoid appearance (Fig. 5A) [1]. Type 2 CRCs demonstrate the worst prognosis among the five molecular subtypes [74,75]. The molecular features of type 2 CRCs (CIMP+/MSI−/BRAF mutation) almost exactly match those of MLH1-unmethylated SSLs and proximal-located TSAs, and these serrated lesions can be precursors of type 2 CRCs (Fig. 5A). As described above, MrTSAs can also be considered precursors of type 2 CRCs. Type 3 CRCs are associated with poor prognosis, although their survival is slightly better than that of type 2 CRCs [74,75]. All subtypes of serrated lesions which frequently harbor KRAS mutations, including sTVAs and SuSAs (both can be classified as USA, as described above) as well as distally located TSAs, can be major precursors of type 3 CRCs (Fig. 5A). KRAS mutations are found in a subset of conventional adenomas, which can also be precursors of type 3 cancers (Fig. 5A). Type 4 carcinomas represent the most common CRC subtype developed through the classic adenoma-carcinoma sequence and are molecularly characterized by chromosomal instability. These cancers typically progress from conventional adenomas, including tubular adenoma, villous adenoma, or TVA (Fig. 5A).
Interestingly, SSLs are thought to be the main precursors of either the best or the worst prognostic CRCs (type 1 and type 2, respectively) (Fig. 5A). Although type 1 and type 2 CRCs commonly share CIMP+/BRAF-mutated status, obtained mainly through the SSL pathway, their contrasting survival rates critically depend on the presence or absence of MSI (that is to say, the presence or absence of MLH1 methylation) [76]. If an SSL harbors MLH1 methylation, the lesion is at a high risk of progressing into an advanced lesion, but an invasive carcinoma derived from the SSL will be expected to show a favorable prognosis. On the other hand, CIMP+ SSLs without MLH1 methylation can be regarded as a potential high-risk precursor of poor-prognostic type 2 CRC. Thus, we believe that for early prevention of poor-prognostic CRCs, it will be helpful to screen CIMP+ SSLs without MLH1 methylation (or CIMP+ SSLs without MLH1 loss) among non-dysplastic SSLs. Collectively, we propose a two-step screening method to stratify risk subgroups of nondysplastic SSLs: first-step screening by combining age, location, and lesion size profiles, and second-step screening by combined molecular and immunohistochemical analyses. This screening approach can efficiently and differentially detect high-risk precursors of type 1 and type 2 CRCs (Fig. 5B). Using this approach, non-dysplastic SSLs at high risk for dysplastic/carcinomatous progression may be more precisely detected. Ultimately, the risksubgrouping of non-dysplastic SSLs will help to prevent interval colon cancers arising from underestimated SSLs.
FUTURE DIRECTIONS
Although notable data refining the morphologic classification and molecular characterization of serrated colorectal lesions have accumulated during the last decade, several pathologic issues remain unresolved. First, overuse of the USA category should be avoided, and USAs should be more clearly divided into morphologic and/or molecular subtypes such as sTVAs and SuSAs. Second, in order to achieve personalized treatment and precision surveillance of serrated premalignant lesions, SSLs and TSAs should be further stratified into risk subgroups based on combined clinicopathologic and molecular profiles; our proposed algorithm for the risk-subgrouping of non-dysplastic SSLs is shown here (Fig. 5B). Third, immunohistochemical or molecular biomarkers should be further developed and validated to aid in the differential diagnosis or prognostic subgrouping of serrated colorectal lesions. Recently studied proteins such as AGRN and LEFTY1 are potential biomarkers which can aid in the differential diagnosis of serrated lesions [62,73].

 XML Download
XML Download