

 PDF
PDF Citation
Citation Print
Print



Recent studies have reported the promising results of immunotherapy in many solid tumors [1]. Unlike traditional cancer therapy, which targets the tumor directly, immunotherapy offers a different approach and is an alternative treatment option for patients with cancer. Because immunotherapy generally engages immune reactions to recognize and eliminate tumor cells, demand for understanding the tumor immune response has increased. Resulting from increased study, novel biomarkers associated with the tumor immune reaction have emerged. These biomarkers may allow innovative approaches in patient selection for immunotherapy and lead to advanced treatment response, expanding the potential impact of immunotherapy. After the advent of U.S. Food and Drug Administration (FDA)–approved anti–programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) therapy agents, the importance of immunotherapy in gastric cancer (GC) has continuously increased. In this review article, we recapitulate the general concept of immuno-oncology and discuss its clinical application while focusing on historical and current clinical trials.
TUMOR IMMUNE MICROENVIRONMENT: GENERAL CONCEPTS AND PROGNOSTIC SIGNIFICANCE
Tumor immune response
While the immune response to tumor cells is mainly caused by CD8-positive cytotoxic T cells, it is a multistep process (socalled cancer-immunity cycle) [2,3], including (1) tumor antigen production by tumor cells and processing of released tumor antigens by antigen presenting cells (APCs) such as dendritic cells (DCs), (2) tumor antigen presentation on the surface of APCs through major histocompatibility complex (MHC) class I and MHC class II substances for recognition by T cells, (3) priming and activation of T cells in lymph nodes after recognition of tumor antigens, (4) migration of activated T cells to tumor through blood vessels, (5) infiltration of T cells into and around the tumor, (6) T cell recognition of antigen presenting tumor cells by binding between MHC class I substances on tumor cells and T cell receptors (TCRs) on the surface of T cells, and (7) killing of tumor cells recognized by T cells. The tumor cells destroyed by T cells release additional tumor antigens, increasing immune response. Some receptors and their ligands have been reported to promote or inhibit each step.
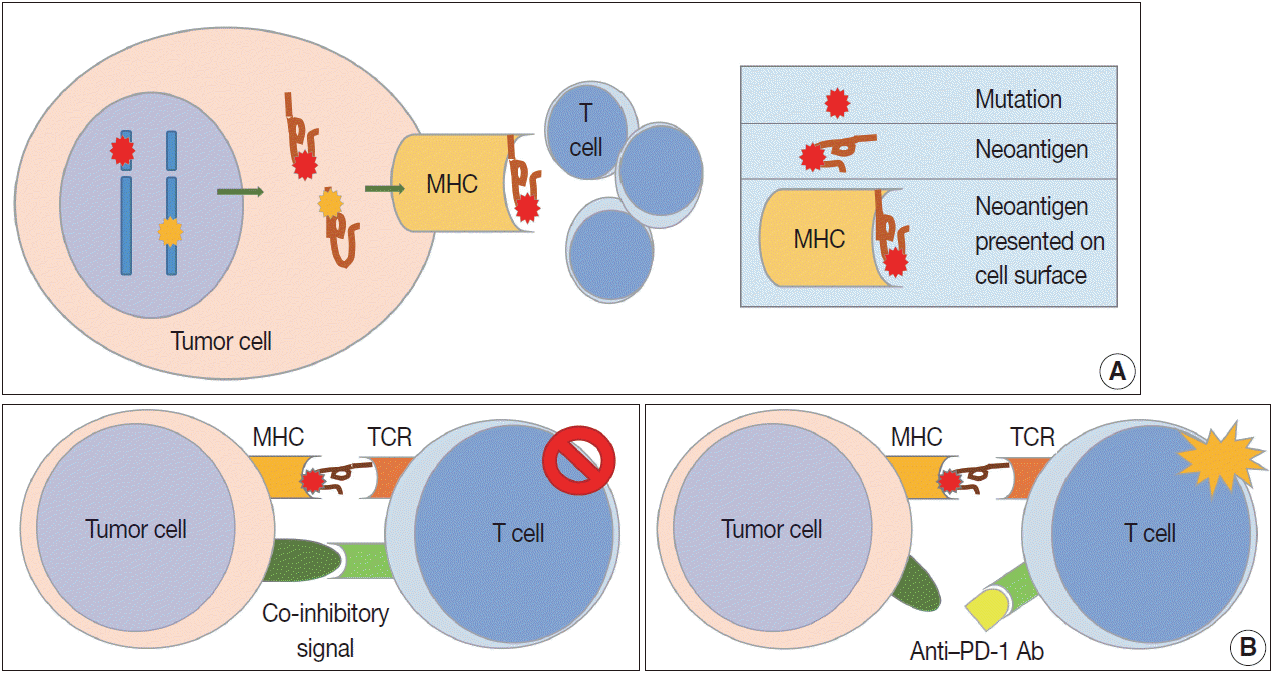
Because patients with tumors have enhanced immune reactions, many researchers have tried to identify tumor antigens that are expressed in tumor cells [4]. Proteins made from gene mutations can be recognized as tumor antigens. As normal cells become tumor cells, mutations are observed in various oncogenes and tumor suppressor genes (Fig. 1A). Furthermore, mutations can easily occur during the gene replication process of cell division due to the genetic instability of tumor cells [5]. Many proteins made from mutated genes are recognized as tumor antigens by immune cells regardless of their function. Neoantigens produced by genetic mutations can induce strong tumor immune responses [6]. Viral proteins from oncogenic viruses, such as human papillomaviruses and Epstein-Barr virus (EBV), are also tumor antigens. In patients infected with the virus, immune cells recognize proteins produced by the virus as antigens, resulting in an immune response to remove the virus-infected cells. Thus, proteins produced from viral genes in tumor cells are important tumor antigens [7]. In addition, proteins that are not expressed in normal cells can be overexpressed in tumor cells. These proteins could be recognized by the immune system as non-self [8]. Some glycoproteins and glycolipids expressed on the cellular surface are overexpressed or modified in tumor cells, which can be recognized as tumor antigens. Oncofetal antigens, such as carcinoembryonic antigen, can also cause immune responses with weak intensity and may be used as diagnostic markers.
Cell-mediated immunity is the main response to tumors, and the inhibitory effect of cytotoxic T cells on tumor cells is well known [9]. Cytotoxic T cells express CD8 and CD3, and have TCRs that recognize tumor antigens presented by MHC class I molecules [10]. T cell proliferation and activation in tumor tissues requires stimulation, usually by two stimuli (Fig. 1B). The first stimulus is a tumor antigen presented on MHC molecules. As described above, neoantigens from genetically mutated or viral genes are major tumor antigens that cause tumor antigen-specific T cell responses. Second, co-stimulatory molecules affect T cell proliferation and survival. T cells have co-stimulatory receptors that combine with expressed co-stimulatory ligands on the surface of tumor cells to promote T cell activation. CD28 and CD27 expression is observed in T cells, and CD134 and CD137 are mainly expressed in antigen-activated T cells [11,12]. Helper T cells are positive for CD4 and CD3 expression. The role of helper T cells in tumor immunity has yet to be fully elucidated, but their main role is to aid humoral immunity via B cell antibody secretion and to enhance the function of cytotoxic T cells [13]. Helper T cells are reported to have cytotoxic functions in some tumors expressing MHC class II. Regulatory T cells (Tregs) express CD4, FOXP3, and CD25 and regulate the immune response by suppressing the immune response to self-proteins and the tumor immune response [14]. Tregs inhibit the tumor immune response by producing high-affinity interleukin-2 (IL-2) receptor, cytotoxic T lymphocyte–associated protein 4 (CTLA-4), IL-10, and immunosuppressive cytokines such as transforming growth factor β (TGF-β).
Mechanism of immune evasion in solid tumors
The immune surveillance system attempts to eliminate tumor cells in the early stages of cancer. However, tumor cells can inhibit the immune system or cause tumor cells themselves to avoid immune responses, a process called immunoediting [15]. Tumor cells can adapt to avoid the immune response by various mechanisms. They can then reach an equilibrium state and escape from the tumor immune response [16].
In contrast to the co-stimulatory receptors described above, tumor cells produce several co-inhibitory receptors, including PD-1, CTLA-4, T cell immunoglobulin, and mucin domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), and indoleamine 2,3-dioxygenase (IDO). These receptors inhibit T cell activation by binding to their tumor cell ligands [11,12]. Tumor-infiltrating lymphocytes (TILS) release interferon γ (IFN-γ) and induce expression of PD-L1 (also known as B7-H1 or CD274) in surrounding tumor cells, stromal cells, and blood cells [17]. Overexpressed PD-L1 activates and binds to PD-1 co-inhibitory receptors on CD8-positive T cells, rendering them inactive. In addition to PD-L1, PD-L2 (also known as B7-DC or CD273) also binds to PD-1 with a weaker binding strength than PD-L1. CTLA-4 is overexpressed early in CD4-positive and CD8-positive T cells. CTLA-4 binds to CD80 (B7-1) and CD86 (B7-2) on APCs, preventing them from binding to the co-stimulatory receptor CD28 and inhibiting TCRs by intracellular PP2A and SHP-2 signaling pathways [18]. CTLA-4 is continuously expressed in Tregs and supports the immunosuppressive function of Tregs [19]. LAG-3 expression is increased in activated T cells and natural killer cells, and its ligands are MHC class II, LSECtin, and galectin-3 [20,21]. LAG-3 inhibits T cell proliferation and cytokine production. TIM-3 binds to its ligands, including galectin-9, high mobility group box 1 (HMGB1), and carcinoembryonic antigen cell adhesion molecule 1, and plays a role in immune evasion of tumor cells by inactivating T cells [22].
MHC molecules are observed on the surface of all nucleated cells, and MHC class I molecules consist of a heavy chain (HLA-A, -B, and -C) and β-2-microglobulin (β2M) [23]. MHC class I molecules play a major role in the cell-mediated immunity caused by cytotoxic T cells. Expression of MHC class I molecules is heterogeneously observed in patients with cancer. Downregulation or loss of MHC class I molecule expression has been reported and is regarded as the main mechanism of avoiding immune responses [24]. β2M gene mutation and alterations in HLA gene transcription and translation are thought to be major causes of decreased MHC class I expression [25].
Tumor cells produce and secrete immunosuppressive factors that inhibit the function of immune cells, such as IL-10, galectins, tumor necrosis factor, TGF-β, prostaglandin E2, and vascular endothelial growth factor [26]. They not only inhibit the function of immune cells but also interfere with their differentiation and maturation. In addition, tumor cells can evade the immune response by modifying tumor antigens [26]. As a tumor grows, tumor cells with immunogenic tumor antigens are removed by the immune response. The immune response can no longer remove tumor cells that lack tumor antigens.
Prognostic significance of tumor-infiltrating lymphocytes in gastric cancer
In recent years, TILs have been studied for their role as prognostic markers and potential therapeutic targets. Neoantigens presented on cancer cells can recruit TILs and trigger an immune reaction. CD8-positive cytotoxic T cells play a role as the effector cytotoxic T cells involved in direct killing of tumor cells [5]. Therefore, they are regarded as the anti-tumorigenic T cell population. Many previous studies reported that higher TIL density was associated with favorable prognosis in patients with cancer, including GC. Furthermore, previous studies consistently demonstrated that higher TIL densities, such as CD3- or CD8-positive cytotoxic T cells, were associated with better outcome in patients with GC [27,28]. These studies investigated TIL density by immunohistochemistry (IHC) of CD3, CD4, CD8, and other markers (Table 1) [29-42]. An image analyzing software was used to quantify TIL density and ensure constant analysis, or TIL density was counted manually. Most studies dichotomized TIL density into a low and high group for statistical analysis. However, the detailed methods and cut-offs are diverse and not yet standardized. For example, analysis areas were selected in various ways and cut-off values for TIL density had a diverse range. TILs were counted in one representative area, two to six representative areas, or in both the center and at the invasive border. Some studies defined cutoffs as a median value, but others defined cutoffs as a mean, 25th percentile, or 60th percentile value calculated in their own cohort. Thus, the cutoff number for CD8-positive cytotoxic T cells ranged from 21.6/mm2 to 946.22/mm2. Although higher TIL density is repeatedly reported as a favorable prognostic biomarker, diagnostic methods are not standardized and there is no consensus regarding the cutoff for high TIL density. Therefore, further study and consensus are needed to clarify the diagnostic reliability and practical usefulness of TIL densities in patients with GC.
CURRENT STATUS OF IMMUNOTHERAPY FOR GASTRIC CANCER
GC is the fourth most common cancer in the world and the second-leading cause of cancer-associated death [43]. For localized GC, the only curative treatment modality is radical surgery with or without perioperative chemotherapy [44]. For unresectable or metastatic GC, systemic chemotherapy has been the mainstay of palliation. Over the last decade, two new agents, monoclonal antibodies targeting human epidermal growth factor receptor 2 (HER2; trastuzumab) and vascular endothelial growth factor receptor 2 (VEGFR-2; ramucirumab), have been approved by the FDA to treat GC in the palliative setting [44]. Briefly, first-line therapy includes a combination of platinum and fluoropyrimidine (5-fluorouracil or capecitabine) with trastuzumab added in HER2-positive tumors [44]. In the second-line setting, therapeutic options include docetaxel, paclitaxel, or irinotecan monotherapy or ramucirumab alone or in combination with paclitaxel [44]. In fact, nearly all patients with advanced GC experience disease progression following treatment [45]. There is currently no standard third-line therapy but options include apatinib, a small molecule multitargeted tyrosine kinase inhibitor with activity against VEGFR-2, and regorafenib, a multikinase inhibitor [44]. More recently, immune checkpoint inhibitors have emerged as among the most advanced therapeutic options available for patients with advanced GC [46].
Pembrolizumab is a selective, humanized, high-affinity IgG4 kappa monoclonal antibody that binds to PD-1, blocking its interaction with PD-L1 and 2 [45,47]. The FDA approved pembrolizumab in May 2017 for patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that progressed even after prior treatment and who have no other optimal treatment choices [48]. The approval was based on promising results of five different clinical trials; the overall/objective response rate (ORR), defined as proportion of patients with a complete or partial response, was 39.6% among 149 patients with various tumor types (95% confidence interval [CI], 31.7% to 47.9%). A complete response was seen in 7% of patients [48]. The duration of response ranged from 1.6+ to 22.7+ months (+indicates no progressive disease at last patient assessment) [48]. The phase 1b KEYNOTE-012 trial had ORR of 22% among 36 patients with PD-L1 positive recurrent or metastatic gastric or gastroesophageal (G/GEJ) adenocarcinomas, and all responses were partial [49]. The phase 2 KEYNOTE-059 trial enrolled 259 patients with locally advanced or metastatic G/GEJ adenocarcinoma to further assess the safety and efficacy of pembrolizumab [50]. The ORR was 11.6% (95% CI, 8.0% to 16.1%), with complete response in 2.3% of cases (95% CI, 0.9% to 5.0%), and the response duration ranged from 1.6+ to 17.3+ months. Among 55% of the tumors with PD-L1 expression based on a combined positive score (CPS)≥1 and that were either microsatellite stable or with undetermined microsatellite instability, the ORR was 13.3% (95% CI, 8.2 to 20.0), with complete response in 1.4% of cases. The duration of response was relatively long, ranging from 2.8+ to 19.4+ months. Based on these results, the FDA granted approval with acceleration for pembrolizumab to treat patients with recurrent, locally advanced or metastatic G/GEJ adenocarcinoma with disease progression on or after two or more standard systemic therapies and if tumors express PD-L1, as determined by an FDA-approved test (PD-L1 IHC 22C3 pharmDx test, Dako, Agilent, Santa Clara, CA, USA) [47]. In the phase 3 KEYNOTE-061 trial, pembrolizumab did not demonstrate significant improvement in overall survival (OS) compared to paclitaxel as second-line therapy for advanced G/GEJ cancer with PD-L1 CPS≥1 [51]. However, pembrolizumab had a better safety profile than paclitaxel. The phase 3 KEYNOTE-062 trial enrolled 763 patients with PD-L1–positive, HER2-negative, advanced G/GEJ cancer; pembrolizumab used as a first-line therapy resulted in noninferior OS compared with standard chemotherapy [52]. Additionally, pembrolizumab showed significant improvement in OS among patients that had tumors with PD-L1 CPS ≥10.
Nivolumab, a human IgG4 monoclonal antibody that inhibits PD-1, has been approved for monotherapy and combination therapy in metastatic melanoma, non-small cell lung cancer, and renal cell carcinoma [46]. The phase 1/2 CheckMate-032 trial showed ORR of 12% in the nivolumab alone group in patients with chemotherapy-refractory G/GEJ cancer [53]. The phase 3 ATTRACTION-2 trial demonstrated significantly longer OS in the nivolumab alone group vs placebo (5.26 months vs. 4.14 months) in patients with advanced G/GEJ cancer refractory to at least two previous chemotherapies [45]. As a consequence, nivolumab was approved to treat advanced GC as a third-line therapy in Japan [46]. In addition, interim results of the phase 2 ATTRACTION-4 trial showed that nivolumab combined with standard chemotherapy showed promising efficacy as a first-line therapy for unresectable advanced or recurrent HER2-negative G/GEJ cancer [54]. The ATTRACTION-4 trial has proceeded to part 2 (phase 3) to compare nivolumab plus standard chemotherapy versus placebo plus standard chemotherapy. Another phase 3 trial (CheckMate-649) is ongoing to evaluate the efficacy and safety of nivolumab combined with standard chemotherapy or ipilimumab (anti-CTLA-4) as a first-line therapy in patients with advanced G/GEJ cancer [55].
There are many other ongoing clinical trials to evaluate the safety and efficacy of a variety of immune checkpoint inhibitors in patients with advanced G/GEJ cancer. These include tremelimumab (anti-CTLA-4) [56], avelumab (anti-PD-L1) [57,58], durvalumab (anti-PD-L1) [56,59], and relatlimab (anti-LAG3) [60]. Most of the trials are currently ongoing. A completed trial (JAVELIN Gastric 300) showed that treatment of patients with G/GEJ cancer with single-agent avelumab in the third-line setting did not demonstrate improved OS or progression-free survival (PFS) compared with standard chemotherapy [57]. The completed and ongoing immunotherapy clinical trials in locally advanced, recurrent, or metastatic G/GEJ cancers are summarized in Table 2.
BIOMARKERS FOR IMMUNOTHERAPY IN GASTRIC CANCER PATIENTS
IHC of PD-L1
PD-1 was first discovered in 1992 by Honjo and colleagues [61]. PD-1 is mainly expressed on activated cytotoxic T cells and other immune cells [51,62]. Within the tumor microenvironment, PD-L1 and PD-L2 can upregulate their expression in cancer cells [62]. In several previous studies and currently published genomic profiles, PD-L1 expression has been observed in more than 40% of GC; it is particularly specific for EBV-positive and MSI-H subtypes [37,63-65].
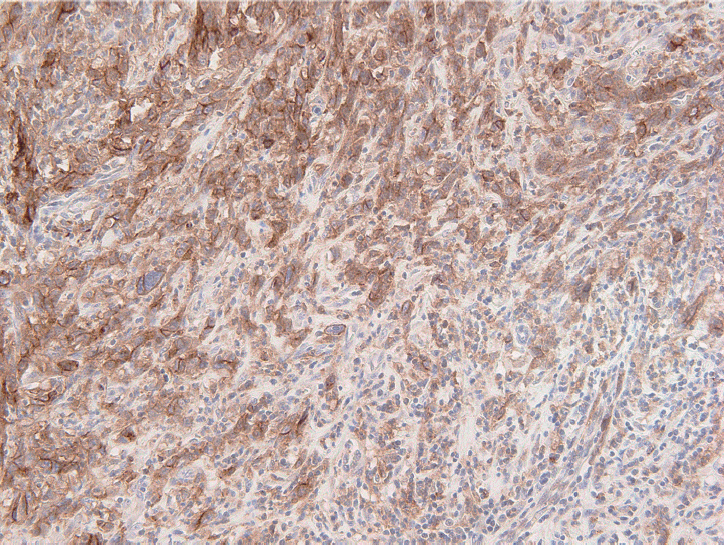
The PD-L1 IHC 22C3 pharmDx assay on the Autostainer Link 48 platform is the FDA-approved companion diagnostic assay to help identify recurrent or metastatic G/GEJ adenocarcinoma in patients who could be treated with pembrolizumab [49,50]. PD-L1 expression assessed by this assay can be quantified by the CPS method (Fig. 2), which is the number of PD-L1 staining cells (tumor cells, lymphocytes, and macrophages) divided by the total number of viable tumor cells, multiplied by 100. If the result of the calculation exceeds 100, the maximum score is regarded as CPS 100 [66]. A tumor with CPS≥1 score is considered positive for PD-L1 expression. For adequate evaluation, at least 100 viable tumor cells are needed in a stained slide [66]. At 20× magnification, partial or complete membrane staining of viable tumor cells and membrane and/or cytoplasmic staining of mononuclear inflammatory cells within tumor and peri-tumoral areas were evaluated, respectively [66]. Another interpretation of PD-L1 expression is the tumor proportion score (TPS), which is evaluation of membrane staining of PD-L1 expression on tumor cells. The Asian ATTRACTION-2 study, which determined the efficacy of nivolumab in advanced G/GEJ adenocarcinoma, retrospectively evaluated PD-L1 expression by PD-L1 IHC 28-8 pharmDx assay (Dako, Carpinteria, CA, USA); PD-L1 positivity was defined as TPS ≥1 [46]. Of note, the ATTRACTION-2 study showed that nivolumab had significant benefit in all patients, regardless of PD-L1 expression [46]. The PD-L1 IHC 28-8 pharmDx assay gained FDA approval as a complementary diagnostic test for nivolumab as second line treatment for advanced non-small cell lung cancer (NSCLC) [67].
Microsatellite instability
MSI-H GC was proposed as a distinct subgroup of GCs by two large scale molecular studies, The Cancer Genome Atlas (TCGA) and the Asian Cancer Research Group classification [65,68,69]. The incidence of MSI-H GCs varies between countries and ethnicity, ranging from 5.6 to 22.7% [65,68,70-73]. MSI-H GCs are associated with antrum location, female gender, relatively older age, earlier stage, and Lauren intestinal type [72]. The presence of dMMR results in tumor cells accumulating frequent genetic mutations. With high mutational burden, tumor cells produce several neo-antigens that trigger T cell activation and recruitment. As the tumor immune reaction increases, expression of checkpoint molecules in tumor cells and immune cells is upregulated [69]. MSI-H GCs are significantly related to both high TILs and high expression of PD-L1 [74,75]. This has been confirmed by several previous genomic profiling studies [76]. On the basis of these hypotheses, recent studies have suggested that MSI-H status may be used as a biomarker for treatment selection with immune-checkpoint inhibitors [50,69]. In Korean patients with metastatic and/or recurrent GC, almost all patients with MSI-H GCs achieve a dramatic response with pembrolizumab alone [75].
Because accurate and reliable evaluation of MSI status is important for treatment decisions, more sensitive, more precise, and faster techniques are required [69]. Three representative methods are currently used to detect GCs with dMMR: polymerase chain reaction (PCR), IHC, and next-generation sequencing (NGS). First, PCR amplification with specific primers for microsatellite repeats is a conventional method using capillary gel electrophoresis [77]. By comparing the allelic position of a microsatellite locus in tumor tissue and corresponding normal tissue, MSI can be determined by peak patterns with a shift [69,77]. The National Cancer Institute recommends the Bethesda Panel, which is composed of five microsatellite markers specific for two mononucleotide repeats (BAT25 and BAT26) and three dinucleotide repeats (D5S346, D2S123, and D17S250) [69,77,78]. Another MSI test is composed of a pentaplex panel of quasimonomorphic mononucleotide markers (NR-27, NR-21, NR-24, BAT-25, and BAT-26) [77,79]. MSI-H is designated when a size shift is identified in at least two of five microsatellites loci [77]. Second, IHC is assessed by expression or total loss of mismatch repair (MMR) proteins (MLH1, MSH2, PMS2, and MSH6) [77]. Loss of expression in one or more MMR proteins is defined as dMMR. Compared with the PCR-based method, IHC has high sensitivity and specificity (more than 90%, respectively) [77,80], but a combination of both methods increases sensitivity. Although IHC allows the determination of which MMR gene is defective, GCs with MSI-H are mainly caused by epigenetic inactivation of MMR genes (hypermethylation of CpG island methylator pathway and hMLH1 silencing) [65,68,72,73]. Therefore, loss of MLH1 and PMS2 was predominantly found in sporadic GCs (~95.8%), unlike with Lynch syndrome [72]. Interestingly, BRAFV600E mutation has not been found in MSI-H GCs, unlike in MSI-H sporadic colorectal cancer [69,81]. Recently developed NGS-based methods cover a broader range of microsatellite loci. However, NGS has several disadvantages: (1) high investment costs per sample; (2) longer run time; and (3) experiential bioinformatics analysis [69,82].
Epstein-Barr virus
EBV-positive GCs constitute a unique subgroup of GCs in the TCGA with several distinct clinicopathologic characteristics, including abundant TILs, male predominance, relatively young age, earlier stage, and favorable prognosis [65,72,73,76]. The incidence of EBV-positive GCs also varies with country and ethnicity, with a range of 2%–20.1% and a worldwide average of nearly 10% [65,72,73,77,83,84]. EBV-positive GCs display CDKN2A (p16INK4A) promoter hyper-methylation, and PD-L1/L2 expression was elevated in genomic profiling, in which IL-12–mediated signaling signatures induced robust presence of immune cells [65,85]. Nearly 50% of EBV-positive GCs showed high expression of PD-L1 [86]. In Korean patients with metastatic and/or recurrent GCs, the patients with EBV-positive GCs achieved dramatic response with pembrolizumab alone [75]. This study suggests that EBV-positive GCs may be good candidates for pembrolizumab monotherapy [75]. Another open-label, multi-arm phase II trial (NCT02951091) is testing the efficacy of nivolumab in EBV-positive GCs as second-line treatment [87]. EBV-positive GCs are commonly accompanied by more extensive infiltration of CD8-positive cytotoxic T cells and IFN-γ, which induce expression of IDO, a potent immune cell inhibitor [76,85]. The IFN-γ driven gene signature, an additional proposed marker of sensitivity to anti–PD-1 treatment, was enriched in EBV-positive GCs [76]. EBV infection demonstrates four latency patterns depending on combinations of latent gene products during the EBV latency cycle: latency Ia, Ib, II, and III. EBV-positive GC demonstrates the latency I pattern, which is limited to EBV-encoded small RNAs (EBERs), BamHI-A rightward transcripts, and Epstein-Barr nuclear antigen 1 [87,88]. The presence of latent membrane protein (LMP) 2A can distinguish latency type Ia or Ib, and LMP2A is expressed in over 50% of EBV-positive GCs [87].
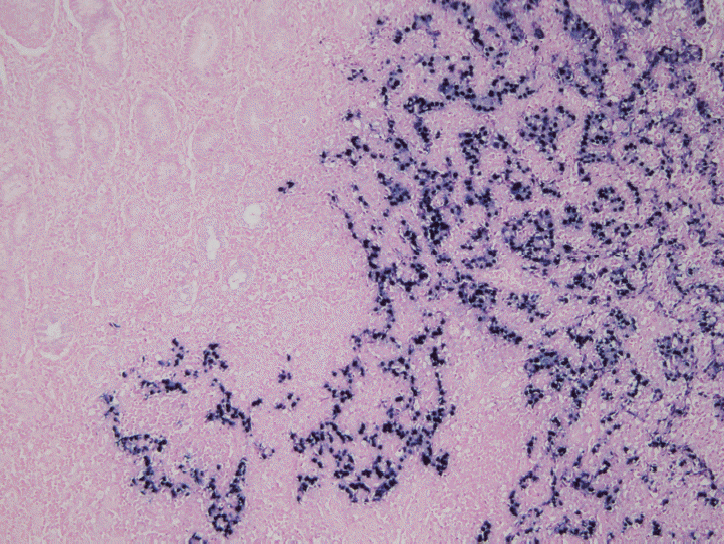
In situ hybridization (ISH) detection of EBER in tumor cells is considered the gold standard to identify the presence of EBV in tissue samples (Fig. 3) [77,89]. EBER ISH allows examination of paraffin sections from surgical excision and biopsy specimens or on cytology preparations [77]. This method uses light microscopy to demonstrate EBER transcripts concentrated in the nuclei of latently infected cells. Cells are considered EBV positive if brown or blue staining is detected in GC cell nuclei. Commercially available EBER probes are labeled with biotin, digoxigenin, or fluorescein (Ventana Medical Systems, Tucson, AZ, USA; Dako, Glostrup, Denmark; Enzo Diagnostics, Farmingdale, NY, USA; Kreatech Diagnostics, Amsterdam, The Netherlands; Novocastra Laboratories Ltd., Newcastle, UK; Shandon Lipshaw, Pittsburgh, PA, USA; Innogenex, San Ramon, CA, USA) [77,90]. Other methods include PCR or serology comparing EBV nucleic acid positivity or sero-positivity in GC tissues versus non-tumor tissues or in blood of GC patients versus healthy controls [91]. Although IHC is easy and convenient, LMP-1 of representative EBV markers is undetectable in EBV-positive GCs [77].
Tumor mutational burden
Tumor mutational burden (TMB), a new predictive biomarker for response to immunotherapy, is a quantitative measure of the total number of somatic nonsynonymous mutations per megabase of genome examined in the DNA of cancer cells [92]. Accumulating data have shown that TMB, or mutational load, is associated with good response to immunotherapy and improved survival [92-96]. Tumors with higher TMB are hypothesized to be more likely to express neoantigens that can be recognized by the immune system in response to immune checkpoint inhibitors [92,97]. The recent CheckMate 227 trial showed that PFS increased in patients with NSCLC treated with nivolumab plus ipilimumab as first-line chemotherapy in tumors with high (≥10 mutations per megabase [Mb]) TMB, regardless of PD-L1 expression [96]. According to this trial, TMB is a promising biomarker for predicting immunotherapy and could lead the way for immuno-oncology entering the era of precision medicine [97]. Clinical trial NCT02915432, which investigated the safety and efficacy of toripalimab in Chinese advanced GC patients, demonstrated that patients with high TMB showed significant treatment response and OS benefit compared with patients with low TMB [98].
TMB can be measured by whole-exome sequencing (WES) or targeted sequencing panels using NGS [92]. Standardized and stabilized sample processing and methodology are crucial factors that influence the reproducibility of TMB measurements [92]. Initial TMB measurements were assessed by WES using tumor tissue and corresponding non-tumor tissue. Because most WES protocols require a minimum of 150–200 ng of genomic DNA, a limited amount of DNA in small biopsy samples is sometimes problematic [92]. The ability to detect somatic nucleic variants depends on allele frequency within a tumor as well as on sequencing depth [92]. Theoretically, at 50× coverage, if an allele frequency of the specific variant is at least 15%, 95% of single nucleotide variants and short insertions and deletions can consistently be identified [92,99]. Unfortunately, WES has some disadvantages: (1) high cost; (2) extensive analysis; and (3) data management [92]. Gene-targeted sequencing panels using NGS have advantages compared with WES: (1) lower sequencing cost; (2) deeper sequencing despite small amount of DNA; (3) higher sensitivity for mutation detection; and (4) shorter turnaround time [92]. Chalmers et al. [100] demonstrated that comprehensive genomic profiling with the FoundationOne assay targeting ~1.1 Mb of the coding genome can accurately evaluate TMB compared to WES and has high correlation. Some commercial gene panels are now available, such as the Ion Torrent Oncomine Tumor Mutation Load Assay (Thermo Fisher Scientific, Waltham, MA, USA), which targets 1.7 Mb of the genome in 409 key cancer genes.
BEYOND PD-1/PD-L1: NEW TARGETS OR COMBINATION THERAPY
While anti–PD-1/PD-L1 therapy has proved successful in GC, many patients do not respond to this treatment or develop resistance. To overcome this limitation of current immunotherapy, a number of ongoing studies are investigating new biomarkers and future strategies for GC treatment. Recently, new immune checkpoints such as LAG-3, TIM-3, and IDO have been suggested as therapeutic targets.
LAG-3 is a member of the immunoglobulin superfamily that acts as a co-inhibitory receptor expressed on exhausted TILs [101]. It may reduce T cell responses by interacting with MHC class II on antigen presenting cells. A previous study by Woo et al. [102] showed that LAG-3 expression is upregulated on TILs, inhibiting T cell immunity and reducing IFN-γ production within the PD-1 upregulated tumor microenvironment. Co-expression of PD-1 and LAG-3 was demonstrated by in vivo study in a murine cancer model, and LAG-3/PD-1 expression was mostly restricted to TILs. This suggests that combination immunotherapy targeting LAG-3 and PD-1 could magnify tumor-specific responses and avoid non-specific autoimmune responses. Several trials assessing the efficacy of combination therapy of immune checkpoint inhibitors are currently ongoing [103]. The FRACTIONGC trial is investigating whether nivolumab in combination with anti–LAG-3 or anti-IDO inhibitor is more effective than nivolumab in combination with ipilimumab (anti–CTLA-4) in treating patients with advanced GC (NCT 02935634). Another trial starting soon will determine the effectiveness of anti–LAG-3 inhibitor+nivolumab alone or in combination with conventional chemotherapy in participants with G/GEJ adenocarcinoma that recurred or metastasized after prior therapy (NCT03704077).
TIM-3 is another co-inhibitory receptor expressed on T cells that induces T cell anergy, apoptosis, and exhaustion through interaction with galectin-9 on immune cells [104]. Additionally, TIM-3 can competitively bind to HMGB1 protein, impairing HMGB1-mediated recruitment of nucleic acids into endosomes, an essential step for DNA sensing by the innate immune system [105]. Because of the T cell exhaustion function, TIM-3 has been suggested as an attractive immunotherapy target [106]. A previous study using murine cancer models showed that combined TIM-3/PD-1 blockade is more effective in controlling tumor growth compared to single blockade of TIM-3 or PD-1 [106]. Another in vivo study confirmed that dual blockade of TIM-3 and PD-1 improved the antitumor function of cancer CD8-positive T cells [106]. An ongoing Phase 1 trial is evaluating the safety of anti-TIM-3 inhibitor administered alone or in combination with anti–PD-L1 antibody in participants with advanced or relapsed solid tumors (NCT03099109).
IDO is a catabolic enzyme produced by Tregs and macrophages to convert tryptophan into kynurenine [107]. IDO-mediated tryptophan deficiency activates naïve T cells and causes them to differentiate into Tregs, causing immune tolerance. In addition, kynurenine converted from tryptophan by IDO also has an immunosuppressive role [108,109]. A study using a melanoma mouse model showed that combinations of CTLA-4 or PD-1/PD-L1 with IDO blockade restored both CD8-positive T cell proliferation and IL-2 production, suggesting the possibility of an IDO inhibitor as a therapeutic option [110]. IDO inhibitors are clinically developing for a variety of cancers, although some trials were stopped due to lack of efficacy [108].
Because of its central role in tumor immunity, HLA molecules could be biomarkers in future immunotherapy. In a previous study of malignant melanoma, down-regulation of the HLA I molecule was suggested as a potential mechanism of resistance to PD-1 blockade therapy [111]. For sophisticated patient selection, confirmation of HLA I molecule downregulation could become essential. A recent study by Chowell et al. [112] reported that zygosity at HLA loci or certain HLA alleles may influence immunotherapy survival. The authors suggested that greater diversity within the HLA I molecule would result in a larger repertoire of neoantigens for presentation. Through HLA genotype data of cancer patients, homozygosity in at least one HLA class I locus was significantly associated with poor prognosis for immunotherapy. Hence, patient HLA genotype might be utilized as a biomarker for immunotherapy discovery.
CONCLUSION
Due to important contributions of past research and clinical trials, immunotherapy is now regarded as an important option for treating patients with advanced GC. The mechanism of the tumor immune reaction is complicated; however, understanding of immune responses to cancer cell is crucial for advanced patient selection. MSI and EBV status are regarded as significant prognostic markers in patients with GC. Along with PD-L1 IHC, MSI, and EBV status are now clinically utilized as predictive markers for immunotherapy. Several ongoing studies and clinical trials are validating the efficacy of novel immunotherapy targeting immune checkpoint molecules including LAG-3, TIM-3, and IDO. As demonstrated by previous clinical trials, immunotherapy will greatly improve survival of patients with GC in the future. Thus, research in immuno-oncology should continue, including studies investigating novel biomarkers and clinical trials.
 XML Download
XML Download