

 PDF
PDF Citation
Citation Print
Print



Gastrointestinal stromal tumors (GISTs) are the most frequent mesenchymal neoplasms of the gastrointestinal (GI) tract [1], with an incidence reported at 10–15 per million persons [2]. GISTs can be found anywhere within the GI tract, most commonly in the stomach (55.6%) and small intestine (31.8%) [1,2]. The cell of origin and diagnostic criteria were highly debated until gain-of-function mutations in KIT were confirmed in 1998 [3-5]. GISTs have a wide morphological spectrum, and although spindle cell tumors are the most common, 20%–25% of GIST cases present an epithelioid pattern, and some cases show mixed histology. Currently, diagnosis is based on morphology, ancillary tests including immunohistochemistry (95% express CD117 and 98% express DOG1), and molecular profiling [6-10]. Among clinicopathological features, the most universally applicable prognostic factors for GISTs are tumor size and mitotic rate per 50 high power fields (HPF) [1,7,8].
Nearly 80% of GISTs harbor a mutation in KIT, and another 5%–10% of cases have platelet-derived growth factor receptor (PDGFR) mutation [6]. However, approximately 10%–15% of GISTs have no mutation in either of these genes and were previously known as wild-type GISTs [6]. However, the terminology “wild-type GISTs” is progressively being abandoned as mutations have been identified in BRAF and succinate dehydrogenase (SDH) genes [9,10]. In tumors with a KIT mutation, the most commonly affected region is exon 11 (juxtamembrane domain; 70%), followed by exon 9 (extracellular domain; 10%–15%), exon 13 (tyrosine kinase 1 domain; 1%–2%), and exon 17 (activation loop; 1%) [11-14]. Secondary mutations in exons 13, 14, or 17 are usually present in imatinib-resistant patients [15].
Management of GIST patients is currently based not only on clinicopathological features (e.g., risk assessment and tumor stage), but also genetic changes including KIT-, PDGFRA-, and SDH mutations [8]. To our knowledge, however, the detailed characteristics and molecular genetic features of GISTs have not yet been described in the Vietnamese population.
In this study, we conducted a standard clinicopathological risk assessment and evaluated the relationships between expression of Ki-67 and p53 and the clinicopathological features of GISTs in Vietnamese patients. Furthermore, we investigated the common regions of KIT mutation, comprising exon 11, exon 9, exon 13, and exon 17.
MATERIALS AND METHODS
Tissue samples
In a descriptive study design, 155 primary GISTs were collected from patients who underwent surgical resection between 2011 and 2014 at the University Medical Center at Ho Chi Minh City (Ho Chi Minh City, Vietnam). Patients treated with preoperative imatinib were not enrolled in the study. The diagnosis was confirmed based on the histological features and immunoreactivity of CD117.
Clinicopathological data of age, sex, tumor location, tumor size, and tumor stage were recorded. Localized GISTs were those that were confined to the primary organ of origin, and locally advanced GISTs were those with contiguous organ involvement. Morphological features, such as pattern (spindle cell, epithelioid, or mixed) and mitotic activity (per 50 HPFs with a total area of 5 mm2) were evaluated. For risk assessment, tumors were categorized into very low-, low-, intermediate-, and high-risk groups based on the National Institute of Health (NIH) classification criteria [16].
For immunohistochemical analysis, we used archival formalin-fixed, paraffin-embedded (FFPE) tissues from the 155 GISTs. One or two representative tumor blocks from each case were examined using immunohistochemistry.
For KIT genotyping, 100 FFPE tissues were available for KIT mutation analysis. Genomic DNA was isolated from paraffin-embedded tissues and tested at KIT exons 9, 11, 13, and 17.
Immunohistochemistry
Three-micrometer-thick slides were used for immunohistochemical analysis. Primary antibodies for CD117 (1:400, polyclonal, Ventana, Oro Valley, AZ, USA), Ki-67 (1:25, MIB-1 monoclonal, Dako, Carpinteria, CA, USA), p53 (1:50, DO-7 monoclonal, Dako), smooth muscle actin (SMA; 1:2,000, 1A4, monoclonal, Dako), and neuron-specific enolase (NSE; 1:50, VI-H14, Dako) were used to detect protein expression. Immunostaining for CD117, Ki-67, p53, SMA, and NSE was performed using an automated staining machine (VENTANA BenchMark, Ventana) according to the manufacturer’s instructions. For the markers (CD117, SMA, and NSE), immunostaining was defined as positive if ≥10% of tumor cells were stained and negative if <10% of tumor cells were stained. The labeling index (LI) (%) for Ki-67 and p53 expression was evaluated based on a count of at least 1,000 tumor cells in the highest-density immunoreactivity areas of the positive tumor nuclei. Ki-67 and p53 were defined as positive if the LI was higher than 10% [17] or 5% [18], respectively. Two senior surgical pathologists without knowledge of the clinicopathological features of the patients independently reviewed the immunostaining slides. Interobserver differences were resolved by consensus review using a double-headed microscope after independent review.
KIT genotyping
Genomic DNA was extracted from paraffin-embedded tissues of 100 patients using the ReliaPrep FFPE gDNA Miniprep System KIT (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Amplifications of KIT exons 9, 11, 13, and 17 were performed using TaKaRa Taq HotStart Polymerase (Takara Bio, Shiga, Japan) with primers as previously published [19]. Polymerase chain reaction (PCR) fragments were sequenced and analyzed in both sense and antisense directions using a BigDye Terminator v3.1 KIT on an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Mutations were confirmed through a second independent PCR amplification and sequencing.
Statistical analysis
Associations between clinicopathological features and Ki-67 expression, p53 expression, and KIT mutations were analyzed using the chi-square test and Fisher's exact test. Differences were considered significant at p<.05. All statistical analyses were performed using SPSS software ver. 16.0 (SPSS Inc., IBM, Chicago, IL, USA).
Ethics statement
This study was approved by the Board of Ethics in Biomedical Research at the University of Medicine and Pharmacy at Ho Chi Minh City, Vietnam (approval number: 54/UMP-BOARD; Date: December 22, 2012). Informed consent was obtained from all individual participants included in the study.
RESULTS
Clinicopathological characteristics of GISTs
We included 72 (46.5%) male and 83 (53.5%) female patients in this study, with a male: female ratio of 1:1.2, and the median age at diagnosis was 55 years (range, 15 to 88 years). Tumor size ranged from 1.0 to 29 cm in the largest dimension, with a median size of 6 cm. The GISTs presented in a wide distribution both within and outside the GI tract (Table 1). The most common location was the stomach (52.3%), followed by the jejunum-ileum (27.7%). GISTs were also found outside the GI tract including the omentum (5.8%) and retroperitoneum (3.2%).
Table 1.
Characteristics of 155 patients with GISTs
![]()
Most cases showed spindle cell (72.3%) or epithelioid cell morphology (14.8%), and mixed type accounted for 12.9% of cases (Table 1, Fig. 1A–C). Tumor cells were positive for CD117 (Fig. 1D). Many GISTs were positive for SMA (20%) (Fig. 1E) or for NSE (67.7%) (Fig. 1F). Sixty-seven of 155 cases (43.2%) were classified as high risk, 42 (27.1%) as low risk, 40 (25.8%) as intermediate risk, and 6 (3.9%) as very low risk (Supplementary Table S1). Most tumors located in the jejunum-ileum (55.8%) or the colorectum (44.4%) fell into the high-risk category (Table 2). Of the 14 cases of extra-intestinal GISTs, 13 (92.9%) were classified as the high-risk group.
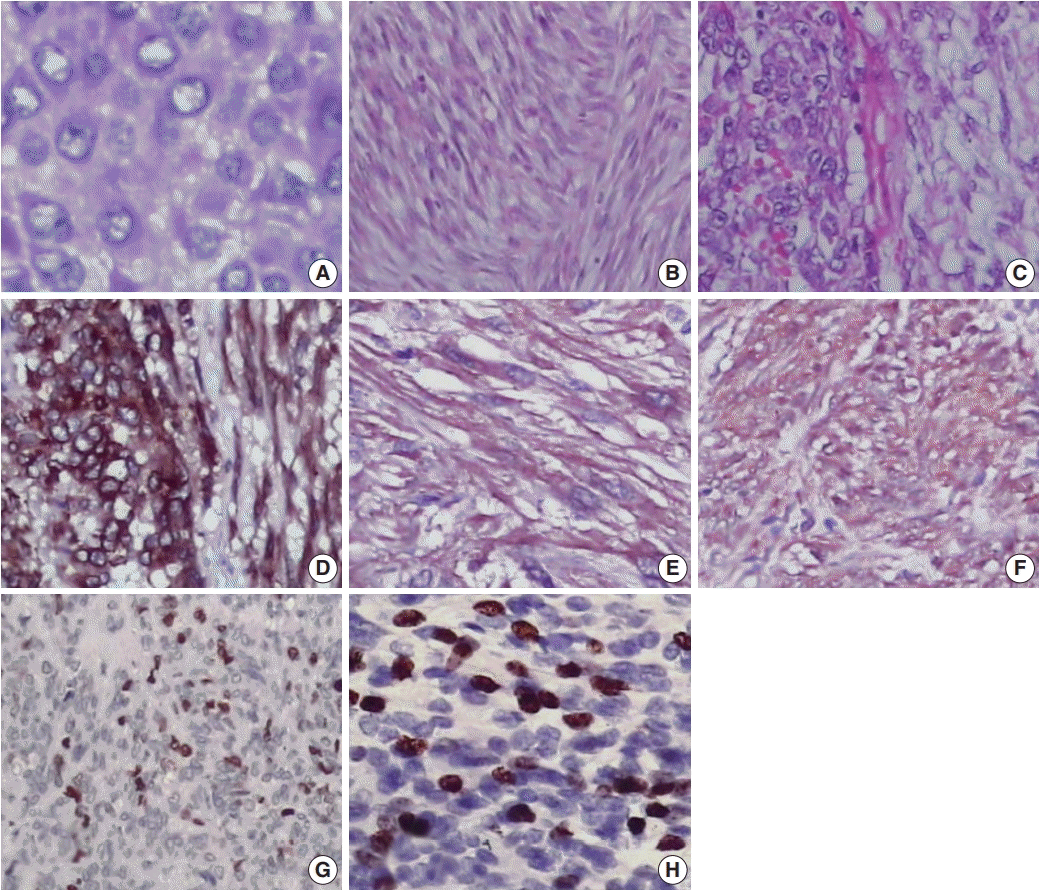
Fig. 1.
Histopathological and immunohistochemical analysis of gastrointestinal stromal tumor. (A) Epithelioid cell morphology. (B) Spindle cell morphology. (C) Mixed type. Immunohistochemical expression of CD117 (D), smooth muscle actin (E), neuron-specific enolase (F), p53 (G), and Ki-67 (H).
![]()
Table 2.
Location and risk stratification of 155 GISTs
![]()
We next evaluated Ki-67 and p53 expression in the GISTs (Fig. 1G, H). Of the 155 cases, 52 (33.5%) were positive for Ki-67, and 58 (37.4%) were positive for p53 (Table 1). Expression of Ki-67 correlated with mitotic rate (p<.001), tumor size (p=.014), risk assessment (p<.001), and tumor stage (p<.001) (Table 3). Similarly, expression of p53 correlated with mitotic rate (p<.001), tumor risk assessment (p<.001), and tumor stage (p<.001). In contrast, expression of Ki-67 and p53 was not associated with GIST phenotype (p=.482 and p=.102, respectively).
Table 3.
Relationship between Ki-67 expression, p53 expression and clinicopathological characteristics of GISTs
|
Ki-67 expression |
p53 expression |
|||||
|---|---|---|---|---|---|---|
| Positive | Negative | p-value | Positive | Negative | p-value | |
| Mitotic rate (per 50 HPFs) | < .001a | .001a | ||||
| ≤ 5 | 19 (20.2) | 75 (79.8) | 25 (26.6) | 69 (73.4) | ||
| 6–10 | 5 (25.0) | 15 (75.0) | 7 (35.0) | 13 (65.0) | ||
| > 10 | 28 (68.3) | 13 (31.7) | 26 (63.4) | 15 (36.6) | ||
| Tumor size (cm) | .014a | .148a | ||||
| 0–5 | 16 (24.2) | 50 (75.8) | 20 (30.3) | 46 (69.7) | ||
| > 5–10 | 17 (32.1) | 36 (67.9) | 20 (37.7) | 33 (62.3) | ||
| > 10 | 19 (52.8) | 17 (47.2) | 18 (50.0) | 18 (50.0) | ||
| Risk stratification | < .001a | <.001a | ||||
| Very low/low risk | 6 (12.5) | 42 (87.5) | 8 (16.7) | 40 (83.3) | ||
| Intermediate risk | 10 (25.0) | 30 (75.0) | 15 (37.5) | 25 (62.5) | ||
| High risk | 36 (53.7) | 31 (46.3) | 35 (52.2) | 32 (47.8) | ||
| Histology | .482a | .102a | ||||
| Spindle | 77 (68.8) | 35 (31.2) | 75 (67.0) | 37 (33.0) | ||
| Epithelioid | 15 (65.2) | 8 (34.8) | 10 (43.5) | 13 (56.5) | ||
| Mixed | 11 (55.0) | 9 (45.0) | 12 (60.0) | 8 (40.0) | ||
| Tumor necrosis | .475b | .012b | ||||
| No | 32 (31.4) | 70 (68.6) | 31 (30.4) | 71 (69.6) | ||
| Yes | 20 (37.7) | 33 (62.3) | 27 (50.9) | 26 (49.1) | ||
| Stagec | < .001b | < .001b | ||||
| Localized | 12 (17.4) | 57 (82.6) | 14 (20.3) | 55 (79.7) | ||
| Locally advanced | 29 (50.9) | 28 (49.1) | 30 (52.6) | 27 (47.4) | ||
![]()
KIT mutation analysis
KIT mutations were detected in 68 of 100 GIST cases (68%). Sixty-two of the 68 KIT mutations (91.2%) were found in KIT exon 11 (Table 4). There were two cases (2.9%) with mutations in KIT exon 9 and 4 cases (5.8%) with mutations in KIT exon 17. No mutations were found in KIT exon 13. We detected a variety of KIT mutation types in exons 9, 11, and 17 including point mutations, insertions, deletions, duplications, and complex mutations.
Table 4.
Summary of KIT mutation status in 100 GIST patients
![]()
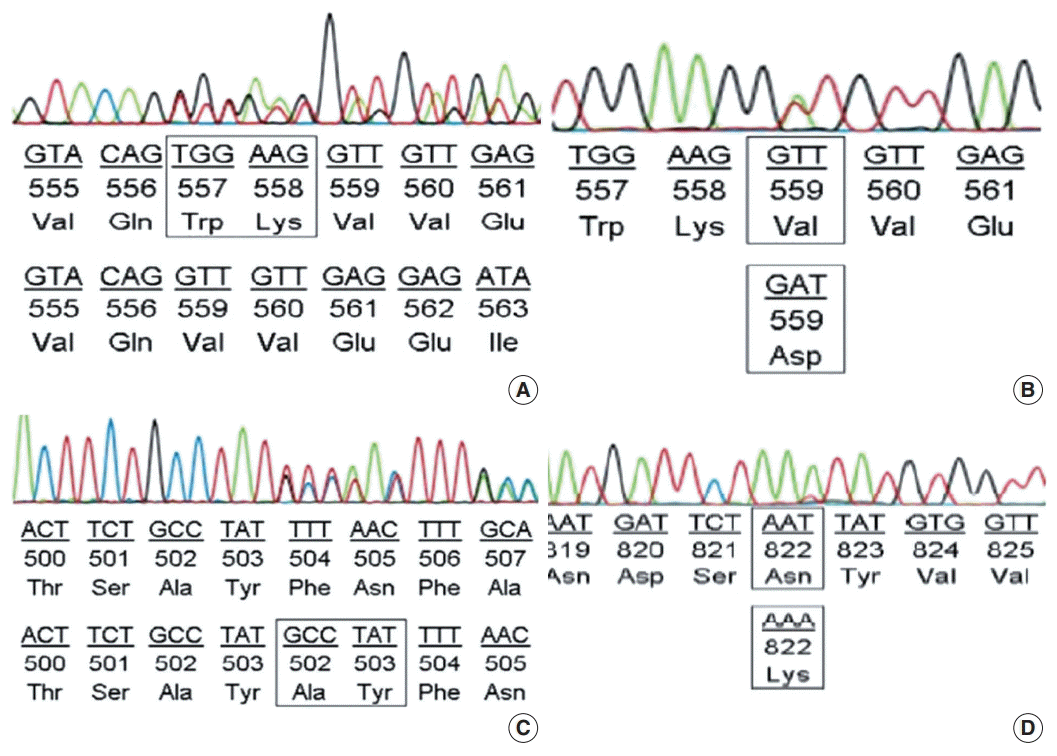
Among the cases with mutations in KIT exon 11, in-frame deletion was the most common type (35/62, 56.5%) (Fig. 2A), followed by point mutation (19/62, 30.6%) (Fig. 2B). Complex mutations were detected in six of 62 cases (9.7%). The two cases with mutation in the extracellular membrane region encoded by exon 9 were insertions (Y503_F504insAY) (Fig. 2C) and were found in tumors of the jejunum-ileum. All mutations in KIT exon 17 were point mutations, and three of four cases (75%) had an N822K substitution (Fig. 2D). We further investigated the relationships between KIT mutation status and other clinicopathological characteristics; however, no significant correlation was found (Supplementary Table S2).
DISCUSSION
A systematic review of 29 studies on nearly 14,000 GIST patients reported a median age of 60 years, and the incidence was similar for males and females [2]. Our study showed a slightly higher incidence rate in females and a median age lower than in other studies. However, our average age was higher than that of the Korean sub-population reported in the systemic review mentioned above [2]. Although many studies have reported the presence of GISTs in the esophagus [1,2], no esophageal GISTs were present in our study. Our results showed that GISTs were most predominant in the stomach, followed by the small intestine, which was consistent with the findings in most of the literature. Interestingly, our results revealed that extra-intestinal GISTs accounted for 9% of cases. According to a study in Singapore from 1998 to 2008, the frequency of extra-intestinal GISTs has increased since CD117 immunohistochemistry has been applied for diagnosis [20].
According to the literature, most GISTs have high-risk features, followed by intermediate- and low-risk groups [2,17,18,20,21]. Our study also had a majority of GISTs classified as high-risk, followed by low-risk, intermediate-, and very low-risk groups. Our study revealed that most jejunum-ileum and colorectal GISTs fell into the high-risk group. These results were compatible with a previous study in which non-gastric GISTs were mostly classified in the high-risk group [22]. Our study showed an extremely high ratio of extra-intestinal GISTs in the high-risk group. In contrast, another study with 29 extra-intestinal GIST cases showed seven cases (24.1%) in the high-risk group using the criteria of mitotic rate per 50 high power fields ≥5/50 HPF and Ki-67 ≥10% [23]. Recently reported evidence proposed that retroperitoneal GISTs could be derived from a GI tract origin [24].
In our study, Ki-67 expression correlated with risk assessment and other clinicopathological features, as in previous studies [17,18,25,26]. Although the cut-off value for Ki-67 expression varied among studies, it is clear that Ki-67 expression is a prognostic factor and a good marker for biological behavior such as metastatic tendency [17,18,25,26]. Furthermore, TP53, the most mutated gene in human cancer [27], has also been proposed to be a predictive marker for risk of malignancy in GISTs [17,28]. The cut-off value for p53 expression varied among studies [17,18,28]. One study from Japan that used the same cut-off values for p53 expression and risk assessment as our study showed less frequent p53 expression in the high-risk group [18]. However, in our study, there was a significant correlation between p53 expression and high-risk assessment. Pauser et al. [28] reported that expression of p53 was related to epithelioid morphology. However, our study showed that p53 expression was not correlated with the morphologic phenotypes of GISTs.
In the current study, KIT mutations were found in 60%–80% of GISTs and were most frequently detected in exon 11, which is consistent with previous reports [1,4,6,29,30]. Additionally, KIT mutations were not associated with the clinicopathological features of GISTs. According to the National Comprehensive Cancer Network (NCCN) panel, the presence and type of KIT mutation are not strongly correlated with prognosis [8]. However, another study showed that GISTs with KIT exon 11 deletions and high mitotic rates were negatively correlated with recurrencefree survival [31]. In the American College of Surgeons Oncology Group (ACOSOG) Z9001 trial, GIST patients with KIT exon 11 deletion mutations had longer recurrence-free survival than those with other mutation types (genotypes) when treated with adjuvant imatinib [32].
Interestingly, the second most frequent region of KIT mutation in this study was exon 17. Two of four cases with KIT exon 17 mutations were identified in the stomach. Other studies have reported that primary mutations in KIT exon 17 were frequently detected in tumors of the small intestine [11,33], and approximately 1% of newly diagnosed GISTs had detectable mutations in KIT exon 17 [11]. Furthermore, KIT exon 17 mutations as a secondary mutation (including N822K substitution) were often found in cases with acquired imatinib resistance [34,35]. Our study did not detect any mutations in KIT exon 13, and mutations in KIT exon 9 were only identified in two cases. According to the NCCN guidelines, patients with advanced GISTs with KIT exon 9 mutations who are treated with double the standard dose of imatinib have an improved likelihood of response [8].
Our study has several limitations. Because it is a descriptive study, it lacks clinical outcome information for the patients, which would enable us to determine overall and disease-free survival. Furthermore, additional analysis to investigate the status of PDGFRA and SDH mutations in the “wild-type” GIST cases (32%) to reveal a more comprehensive picture of the molecular alterations in Vietnamese GIST patients would be helpful but was not feasible at this point.
In conclusion, this study demonstrated the clinicopathological features, immunohistochemical characteristics, and KIT mutation status of 155 Vietnamese GIST patients. The stomach was the most common site of GISTs, followed by the small intestine, outside the GI tract, and colorectum. The majority of GISTs outside the GI tract fell into the high-risk group. Expression of Ki67 and p53 was associated with high-risk assessment. Mutations in KIT exon 11 were the most frequently detected, followed by mutations in KIT exons 17 and 9. However, KIT mutations were not associated with clinicopathological or morphological features.
 XML Download
XML Download