

 PDF
PDF Citation
Citation Print
Print



Primary breast sarcoma (PBS) is a rare heterogeneous group of tumors, which comprises approximately 1% of breast malignancies [1]. Distribution of histologic subtypes varies among studies; however, angiosarcoma is the most prevalent while liposarcoma, fibrosarcoma, and malignant fibrous histiocytoma have also been reported [2-4].
Rhabdomyosarcoma (RMS) is one of the most common sarcomas in young patients; however, RMS of the breast origin is very rare, with only 26 cases reported in international journals [5-7]. Among the reported cases, alveolar subtype was most common, followed by embryonal, spindle cell/sclerosing, and pleomorphic subtypes. Spindle cell/sclerosing RMS (ssRMS) is a new subtype of RMS included in the World Health Organization Classification of Tumors of Soft Tissue and Bone in 2013 [8], and only one primary breast ssRMS has been reported in an international journal [9].
We recently encountered a case of ssRMS of the breast that could have been misdiagnosed as some other spindle cell sarcoma. To better understand the clinicopathological characteristics of these rare conditions, we searched and reviewed data from Asan Medical Center databases and reviewed the literature related to this disease.
MATERIALS AND METHODS
Case selection
Upon reviewing Asan Medical Center pathology database between January 1, 2000 and November 30, 2018, 41 PBS cases including three cases of primary RMS of the breast were retrieved. Cases of metastatic sarcoma and radiation-induced sarcoma were excluded. The clinicopathological features of primary RMS were analyzed, and literature related to PBS and primary RMS of the breast was reviewed.
Immunohistochemical study
For immunohistochemical (IHC) staining, 4-μm thick tissue sections were deparaffinized and hydrated by immersion in xylene and a graded ethanol series. Endogenous peroxidase was blocked by incubation in 3% H2O2 for 10 minutes, and then heat-induced antigen retrieval was performed. Primary antibody staining was performed using a Benchmark autostainer (Ventana Medical Systems, Tucson, AZ, USA) following the manufacturer’s protocol. Sections were incubated at room temperature for 24 or 32 minutes in primary antibodies for desmin (1:200, D33, Dako, Glostrup, Denmark), myogenic differentiation 1 (1:50, MyoD1, EP212, Cell Marque, Rocklin, CA, USA), myogenin (1:200, Neomarkers, Fremont, CA, USA), smooth muscle actin (SMA; 1:4,000, 1A4, Dako), nestin (1:1,000, 10C2, Cell Marque), CD34 (1:500, QBEND10, Immunotech, Marseille, France), CD56 (1:100, 504, Leica, Chicago, IL, USA), pancytokeratin AE1/AE3 (CK AE1/AE3, 1:400, Leica), and pancytokeratin MNF-116 (1:100, MNF 116, Dako), and then labeled with an iView detection kit using the automated immunostaining system (Benchmark XT, Ventana Medical Systems). Immunostained sections were lightly counterstained with hematoxylin, dehydrated in ethanol, and cleared in xylene.
Nested real time polymerase chain reaction (nested reverse transcription polymerase chain reaction)
An alveolar RMS (aRMS) case was examined for nested reverse transcription polymerase chain reaction (RT-PCR) of FAX3/ TKHR (FOXO1) fusion and FAX7/TKFR (FOXO1) fusion gene transcript. After the histologic and IHC examinations, formalin-fixed paraffin-embedded block were used for the test. DNA was extracted using the RecoverAll Total Nucleic Acid Isolation Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. The DNA quality was assessed by amplification of the housekeeping gene β-globulin.
The nested RT-PCR was carried out with ABI 7500 RT-PCR system (Applied Biosystems, Foster City, CA, USA) using 2 μL of genomic DNA as the template, 1 μL of each primer, 1 μL of each probe, 2 μL of dNTP, 2.5 μL of buffer, 0.1 μL of Taq polymerase, and 16.4 μL of distilled water, in a total volume of 25 μL. The synthesized primer PAX3/PAX7 (5'-CCGACAGCAGCTCTACCTAC-3'), PAX3 (5'-CTACTGCCTCCCCAGCAC-3'), PAX7 (5'-CACAGCTTCTCCAGCTACTCTG-3' and 5'-CACAGCTTCTCCAGCT-3'), and FKHR (5'-GCACACGAATGAACTTGCTG-3' and reverse transcript 3'-CCAAGAACT TTTT CCAGTTCC-5') was used. The thermal cycling conditions of the first cycle of nested RT-PCR were 5 minutes at 94°C, followed by 35 cycles of 40 seconds at 94°C, 40 seconds at 55°C and 1 minute at 72°C. After 1 cycle of 10 minutes at 72°C, second RT-PCR was performed with the same thermal cycling condition. A positive result for PAX3/PAX7-TKHR (FOXO1) was defined as a threshold cycle (CT) value <40, and the internal control was defined as a CT value <36.
Ethics statement
The study plan was reviewed by the institutional review board of Asan Medical Center and it was exempted from deliberation (exemption number: 2018-0883); informed consent was obtained from the only living patient.
Go to : 
RESULTS
Case descriptions
Patient A
Patient A was a 14-year-old Korean girl who had a painful 10-cm mass in her left breast. Magnetic resonance imaging (MRI) revealed a huge, heterogeneously enhancing mass measuring 12×9.5×8.3 cm that involved her whole left breast. No other abnormalities were identified on MRI and computed tomography (Fig. 1A).
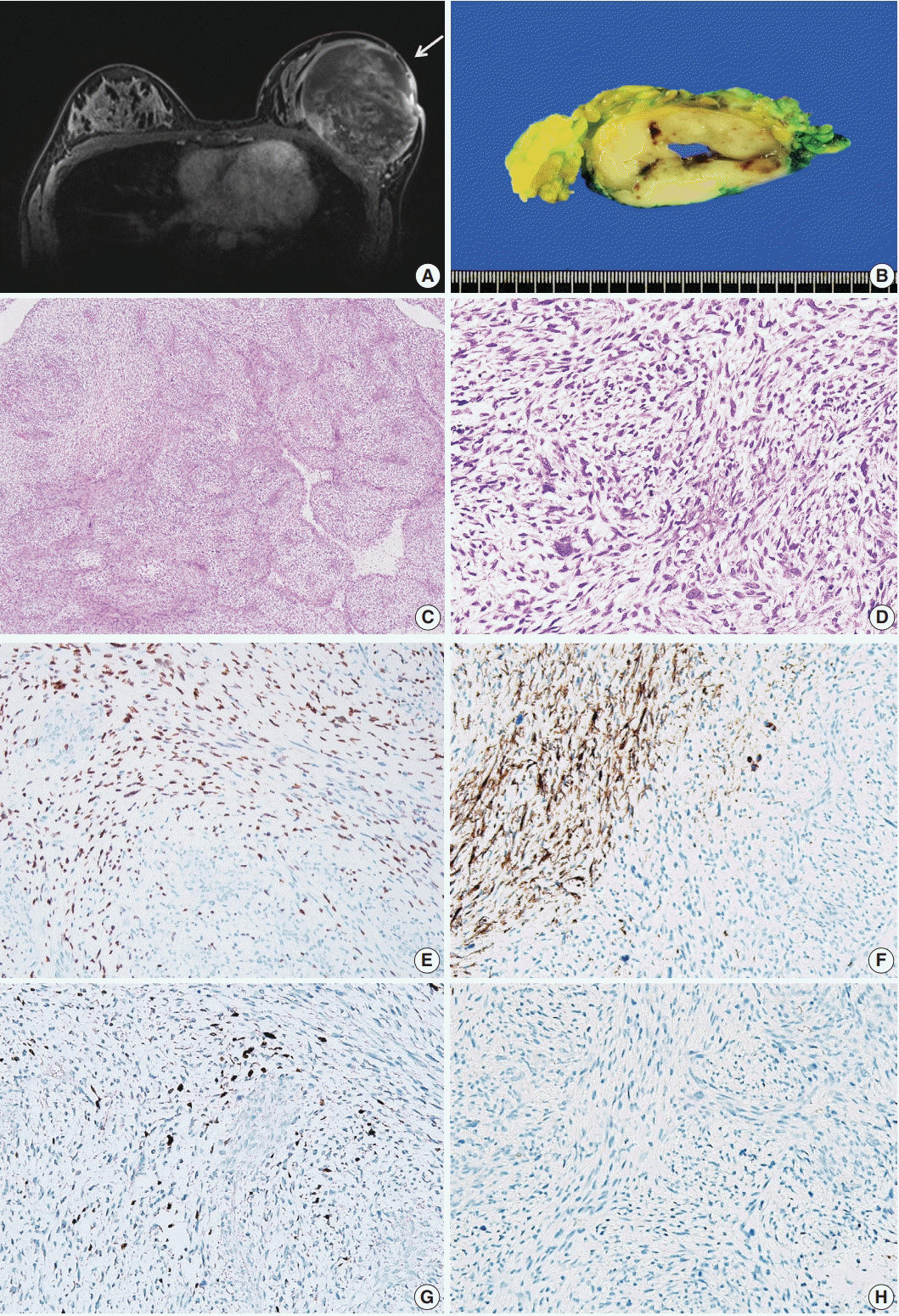 | Fig. 1.Primary breast sarcoma of patient A. (A) A 12×9.5×8.3-cm-sized heterogeneously enhancing mass in the left breast (yellow arrow). (B) Grayish-yellow cut surface of the mass with multifocal hemorrhagic spots and central cavity formation. (C, D) Spindle cells exhibiting a fascicular or storiform growth pattern with a small amount of myxoid matrix. The image showing highly pleomorphic elongated nuclei and pale to eosinophilic cytoplasm, with high mitotic rates (up to 94/10 high power fields). (E–H) Immunohistochemical staining showing positivity of tumor cells for MyoD1 (E), desmin (F), and myogenin (G) and negativity for MNF-116 (H).
|
A skin-sparing mastectomy was performed. The surgical specimen included fragmented tumor tissue measuring 16.7× 10.2×3.5 cm in an aggregate, weighing 711 g. The cut surface of the mass showed creamy white-colored trabecular features with firm texture (Fig. 1B). Microscopic examination revealed a mesenchymal tumor composed of spindle cells, with a fascicular growth pattern and varying amount of myxoid matrix (Fig. 1C). The nuclei were elongated and highly pleomorphic; the cytoplasm was pale to eosinophilic (Fig. 1D). The mitotic rate was extremely high (up to 94/10 high-power fields). No epithelial component was identified in the tumor. On IHC staining, the tumor cells tested positive for desmin, MyoD1, myogenin, and nestin but not for CK AE1/AE3, MNF-116, SMA, CD34, and S100 (Fig. 1E–H). ssRMS of the breast was diagnosed based on histological and IHC findings. Resection margin could not be assessed because of the disruption caused during mastectomy. She was postoperatively treated with combination chemotherapy (vincristine, actinomycin, and cyclophosphamide).
Four-month post-mastectomy, a mass was identified at the operation site and diagnosed as a recurrent ssRMS. The patient continued chemotherapy (cyclosphosphamide, doxorubicin, and ifosfamide) and radiation therapy.
Patient B
Patient B was a 16-year-old Korean girl who had a 10-cm mass in her left breast. Imaging examination at an outside hospital revealed a breast mass and multiple metastatic lesions in the lumbar spine and left axillary lymph nodes. She underwent core needle biopsy of the breast mass; the tumor was diagnosed as aRMS.
After receiving the first neoadjuvant chemotherapy cycle, she was referred to our center and continued to receive the treatment for 8 cycles. However, MRI showed no change in the status of the left breast and metastatic masses.
The patient underwent palliative mastectomy. The surgical specimen consisted of a lump of breast tissue that measured 9.5× 8.5×4 cm and weighed 114 g. A well-demarcated lobulating soft mass measuring 4×2.5×2 cm was identified in the breast parenchyma; the resection margins were clear. The mass was composed of nests and sheets of primitive round cells separated by fibrous septa. These nests exhibited a central loss of cellular cohesion (Fig. 2A). IHC staining of the tumor cells yielded positive results for desmin (Fig. 2B), CD56, and myogenin (Fig. 2C) and negative results for CK AE/AE3 and SMA.
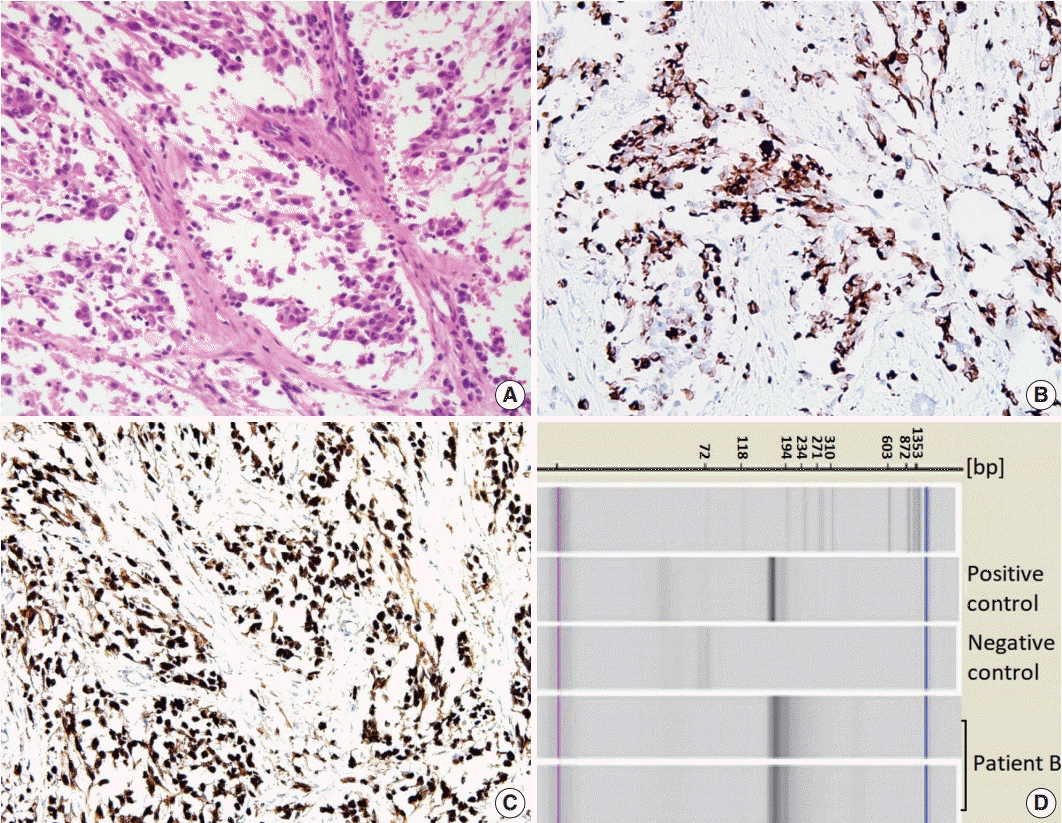 | Fig. 2.Primary breast sarcoma of patient B. (A) Primitive round-to-polygonal cells comprising the nests separated by fibrous septa and showing a loss of cellular cohesion. (B, C) Immunohistochemical staining for desmin (B) and myogenin (C). (D) PAX3-FKHR (FOXO1) fusion identified by PAX-3/PAX-7-FKHR (FOXO1) nested reverse transcription polymerase chain reaction analysis.
|
PAX3-FKHR (FOXO1) fusion transcripts [t(2;13)(q35;q14)] were identified using nested RT-PCR, supporting the diagnosis of aRMS (Fig. 2D). Post-surgery, she received eight chemotherapy cycles (carboplatin, VP-16, and ifosphamide), which was interrupted by fungal infection and low platelet counts. Followup imaging examination revealed recurrence of the chest wall mass and exacerbation of the multifocal bone metastasis with bone marrow involvement. The patient died due to respiratory suppression 22-month post-diagnosis.
Patient C
Patient C was a previously healthy 25-year-old Korean woman who had palpable masses on her left breast. Ultrasonography and MRI revealed multiple infiltrative masses in her left breast that measured up to 3 cm (Fig. 3A). Positron emission tomography revealed multiple hypermetabolic lesions in the left breast and axillary lymph nodes.
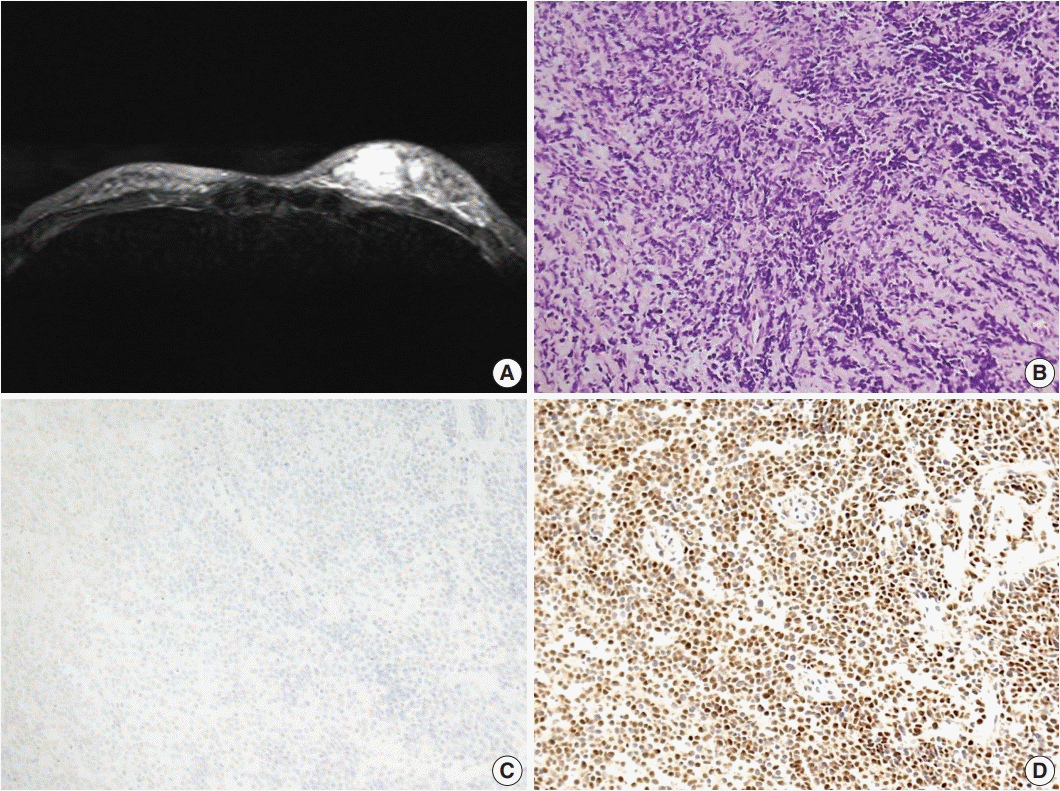 | Fig. 3.Primary breast sarcoma of patient C. (A) Magnetic resonance imaging of the breast showing multifocal, infiltrating, ill-defined, enhancing masses in the left lower inner, upper inner, and lower outer quadrants. (B) Hyperchromatic small round cells with scanty cytoplasm exhibiting solid-sheet and cord-like growth patterns. (C, D) Immunohistochemical staining showing negativity for desmin (C) and positivity for myogenin (D).
|
Ultrasonography-guided needle biopsy of the breast mass was performed. The lesion consisted of loosely cohesive small round cells showing solid sheet or cord-like growth patterns. The cells had scant amounts of cytoplasm and round-to-polygonal shaped hyperchromatic nuclei (Fig. 3B). On IHC examination, these cells showed nuclear positivity for myogenin and negativity for desmin (Fig. 3C, D). The tumors were diagnosed as embryonal RMS (eRMS) based on histological and IHC findings.
The patient received 5 cycles of neoadjuvant chemotherapy (vincristine, adriamycin, cyclophosphamide, ifosfamide, and etoposide) and radiation therapy; however, 8 months post-diagnosis, multiple metastatic lesions were found in bone and left upper lobe of the lung. She received additional chemotherapy but died due to adriamycin cardiotoxicity 18 months post-diagnosis.
Other PBSs
A total of 41 patients were diagnosed with PBS from 2000 to 2018 at Asan Medical Center (Table 1). Among these, two cases were males (5%); one case each of dermatofibrosarcoma protuberans (DFSP) and fibrosarcoma. Mean age of the 41 patients was 44.1 years (range, 14 to 75 years); the most common age group was 50–60 years old (15 cases, 37%) (Table 1).
Table 1.
Distribution of primary breast sarcoma according to age (review of single institution)
![]()
Angiosarcoma was the most common type of PBS (13 cases, 32%), followed by extra-skeletal osteosarcoma (5 cases, 12%), liposarcoma, and DFSP (4 cases, 10%, respectively); other PBS types occurred with a similar frequency as each other (Table 1, Fig. 4). Among patients aged less than 30 years (7/41 cases), angiosarcoma was not the dominant subtype; instead, we identified three cases of RMS and one case each of undifferentiated spindle cell sarcoma, DFSP, angiosarcoma, and leiomyosarcoma (Table 1, Fig. 4).
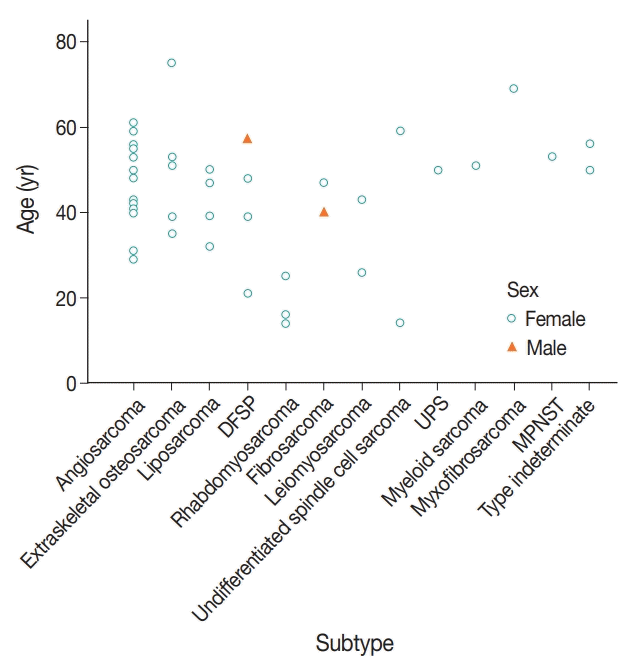 | Fig. 4.Age distribution of primary breast sarcoma (PBS) according to subtype. Angiosarcoma was the most common sarcoma, followed by extra-skeletal osteosarcoma, liposarcoma, and dermatofibrosarcoma protuberans (DFSP). The most prevalent age-group was 50–60 years. PBS was rare in people aged less than 30, and rhabdomyosarcoma was the most common subtype in this age-group. UPS, undifferentiated pleomorphic sarcoma; MPNST, malignant peripheral nerve sheath tumor.
|
Two cases were consultation cases for which only hematoxylin and eosin slides were available for review, and thus it was difficult to confirm the subtype. These were categorized as indeterminate type.
Go to :
DISCUSSION
PBSs are a group of mesenchymal-derived de novo malignancies, accounting for less than 1% of breast cancer [4,10]. Due to their rarity, large scale analyses have been limited, and PBS has no known etiology [4,11]. Some breast sarcomas are related with previous treatment (radiation therapy, therapy-associated chronic lymphedema and variable chemicals), and they are categorized as secondary breast sarcoma [4,11,12].
Thus far, angiosarcoma is the most commonly reported PBS; virtually any type of sarcoma can occur in the breast as a PBS [13]. Based on our 18-year institutional experience, angiosarcoma is the most common type of PBS, followed by extra-skeletal osteosarcoma and liposarcoma. However, the distribution of PBS subtypes is not consistent across age groups; angiosarcomas were mostly found in middle-aged women (age, 40 to 60 years). Among young females (age, 10 to 30 years), angiosarcoma was rare. RMS was the most common subtype of PBS in this age group (Table 1, Fig. 4).
Only 26 cases of RMS of breast origin have been reported in English literature, almost exclusively in the young females [7]. Demographic features, treatments, outcomes, and associated genetic findings of the 29 RMS cases (including our 3 cases) are given in Table 2.
Table 2.
Cases of primary rhabdomyosarcoma of the breast (review of literature)
| Study | Age (yr) | Subtype | Size (cm) | LN | Meta | Surgical procedure | Other treatment | Prognosis |
|---|---|---|---|---|---|---|---|---|
| Hays et al. (1997) [18] | 13.6 | Alveolar | 3 | + | + | NI | RTx | DOD 79 wk |
| 16.9 | Alveolar | 15 | + | + | Excision | CTx, RTx | DOD 42 wk | |
| 15.5 | Alveolar | 21 | + | – | Radical mastectomy | CTx | NED 252 wk | |
| 16.9 | Alveolar | 8 | – | – | Radical mastectomy | RTx | NED 365 wk | |
| 15.2 | Alveolar | 6.5 | – | – | Mastectomy | RTx | NED 150 wk | |
| 15 | Alveolar | NI | – | + | NI | NI | DOD 124 wk | |
| 14.9 | Embryonal | 10 | – | – | Excision | RTx | NED 362 wk | |
| Hererra and Lugo-Vicente (1998) [19] | 13 | Embryonal | 6 | – | – | NI | CTx | NED 1 yr |
| Binokay et al. (2003) [20] | 16 | Alveolar | 10 | + | – | MRM | NI | NI |
| Vishnevskaia et al. (2004) [21] | 14 | Alveolar | NI | + | + | NI | NI | DOD 3 yr 1 mo |
| Italiano et al. (2005) [14],a | 46 | Embryonal | 3.5 | NI | NI | Quadrantectomy | CTx, RTx | NED 18 mo |
| Nogi et al. (2007) [23] | 13 | Alveolar | 13 | + | – | Total mastectomy | CTx | DOD 8 mo |
| Attili et al. (2007) [5] | 40 | Embryonal | 4 | + | – | MRM | CTx | NED 1 yr |
| Rasinariu et al. (2011) [9] | 58 | Spindle cell/sclerosing | 11 | NI | NI | Mastectomy | NI | NI |
| Li et al. (2012) [12] | 30 | Alveolar | 2.5 | + | – | MRM | neoCTx | DOD 29 mo |
| 17 | Embryonal | 10 | – | – | Simple mastectomy | NI | NED 34 mo | |
| Valera et al. (2013) [24] | 17 | Alveolar | 3.1 | NI | + | None | CCRT | NI |
| Bhosale et al. (2013) [25] | 60 | NI | 8 | + | – | MRM | CTx | NED 6 mo |
| Mondal et al. (2014) [26] | 49 | Pleomorphic | 7 | – | – | MRM | None | NED 12 mo |
| Kallianpur et al. (2015) [27] | 19 | NI | 30 | – | – | Mastectomy | CTx, RTx | NED 2 mo |
| Pareekutty et al. (2016) [28] | 12 | Alveolar | 9 | + | – | MRM | CTx, RTx | NED 35 mo |
| Audino et al. (2017) [7] | 16 | Alveolar | 12.5 | + | + | MRM | CTx, RTx | Recur 19 mo |
| Kim et al. (2017) [10] | 17 | Alveolar | 3.1 | – | + | Mastectomy | CTx | Recur 5 mo |
| Yuan et al. (2017) [29] | 34 | NI | 3.5 | NI | NI | Mastectomy | CTx | NED 23 mo |
| Bayramoglu et al. (2018) [30] | 12 | Alveolar | NI | + | – | NI | NI | NI |
| Jean-Louis (2018) [31] | 16 | Alveolar | 8.1 | + | – | Excision | NI | NI |
| Current study | 14 | Spindle cell/sclerosing | 16.7 | – | + | Mastectomy | CTx, RTx | Recur 4 mo |
| 16 | Alveolar | 9.5 | + | + | Mastectomy | CTx | DOD 22 mo | |
| 25 | Embryonal | 3 | + | – | - | CTx, RTx | DOD 18 mo |
LN, lymph node status; Meta, metastasis; NI, not identified; RTx, radiotherapy; DOD, dead of disease; CTx, chemotherapy; NED, no evidence of disease; MRM, modified radical mastectomy; neoCTx, neoadjuvant chemotherapy; CCRT, concurrent chemoradiation therapy.
a This was an identical case of Dausse et al. (2005) [22].
![]()
Among the 29 cases, aRMS was the most common type (n= 17, 59%), followed by eRMS (n=6, 21%), ssRMS (n=2, 7%), and pleomorphic RMS (n=1, 3%). Subtype-related information was not available for three cases (n=3, 10%). All cases involved female patients; median age was 16.4 years (range, 11 to 60 years). Majority of the cases involved adolescents and young adults (range, 10 to 30 years; 24 cases, 83%); five cases (19.2%) involved middle-aged patients. All but one aRMS cases involved teenage girls; contrastingly, eRMS cases were almost equally distributed between adolescents and middle-aged women; the other RMS types mostly involved middle-aged patients. Interestingly, aRMS cases frequently presented with axillary lymphadenopathy, mimicking mammary carcinomas.
To the best of our knowledge, the ssRMS case in this series is the second to be reported in the literature. ssRMS is a newly classified subtype that accounts for 5%–10% of all RMS cases; it mostly involves paratesticular lesions in pediatric populations and head and neck and genitourinary lesions in adults [8]. As ssRMS was defined as a variant of eRMS, we reviewed all reports of eRMS for reclassification; however, none of the eRMS cases had the histological characteristics that indicated ssRMS.
Differential diagnoses vary according to RMS subtype, however, differentiation of malignant phyllodes tumor (MPT) with sarcomatous overgrowth is always important regardless of subtypes [2]. Age can be used to differentiate these malignancies; most MPTs occur in the fifth or sixth decade of life, whereas primary RMS of the breast mainly affects young patients [3,14,15]. Extensive tissue sampling is necessary, especially in middle-aged patients, as the epithelial component of the MPT can be very small [2].
IHC results are critical after excluding MPT with sarcomatous overgrowth; single staining or a combination of desmin, MyoD1, and myogenin staining have been used to diagnose RMS. However, Parham et al. [16] suggested that desmin should always be used as part of a panel and never as a sole diagnostic marker, as it can non-specifically stain other small round cells and smooth muscle cells. However, despite positive IHC results, the possibility of other myogenic neoplasms cannot be excluded in some cases; therefore, genetic analysis is useful for both histological typing and diagnosis [8]. Two characteristic cytogenetic changes— t(2;13)(q35;q14) resulting in PAX3-FHKR (FOXO1) fusion and t(1;13)(p13;q14) resulting in PAX7-FKHR (FOXO1) fusion— are identified in about 80% of aRMS cases, which allow aRMS to be distinguished from other types of round cell neoplasms and RMS [8]. These fusions are known to be associated with the activation of transcription from PAX3/PAX7-binding sites and to contribute to tumorigenesis [17]. In the D9803 COG study, failure-free survival of fusion-positive aRMS patients was lower than that of fusion-negative patients, and PAX3-FKHR (FOXO1) fusion-positive patients showed higher overall survival than PAX7-FKHR (FOXO1) fusion–positive patients [17].
Due to its rarity, definite treatment has not been established for RMS of the breast. Generally, axillary dissection of PBS is not recommended as sarcoma usually does not metastasize to lymph nodes; axillary dissection is only required in cases involving palpable lymphadenopathy. However, more than 50% of breast aRMS cases (12/17 cases, 71%) showed axillary node metastasis; moreover, 58% of these cases had no disseminated metastases (7/12 cases). These findings might suggest that axillary node metastasis of aRMS is not only incidental findings of disseminated metastasis, but also early event of disease spreading. Therefore, axillary node dissection or sentinel node sampling of aRMS needs to be considered in cases that require surgery.
RMS of the breast is a rare malignancy that mainly occurs in young females. Our experience regarding RMS cases suggests that spindle cell or small round cell malignancies in the breasts of young females should be suspected of being primary or secondary RMSs. This is the second report that describes a case of ssRMS of the breast, and it may help clinicians who encounter this rare disease in the future [5,7,9,10,12,14,18-31].
Go to :
 XML Download
XML Download