

 PDF
PDF Citation
Citation Print
Print



Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide, and its incidence is increasing because of the dissemination of hepatitis B and C viruses [1,2]. HCC is the third most common cause of cancer-related deaths, with approximately 600,000 deaths per year [3-6]. Although surgical resection and transplantation are effective treatments for HCC, most patients present with advanced-stage disease at the time of diagnosis and are not suitable candidates for curative surgery [4]. Inoperable HCC patients undergo various loco-regional treatments, including transarterial chemoembolization, radiofrequency ablation, and percutaneous ethanol injection, with or without systemic chemotherapy using adriamycin, cisplatin, or sorafenib [7-9]. Despite the development of various treatments for advanced HCC patients, HCC remains difficult to treat because these methods do not significantly improve mortality [10]. Therefore, novel approaches to develop targeted therapeutic agents are needed.
Immortalized cancer cell lines have been typically used for highthroughput screening of chemical libraries to develop novel therapeutics in the past. Although immortalized cancer cell lines have the advantage of being robust and tractable, they have significant limitations recapitulating tumor heterogeneity of in vivo human tumors [11-13]. Primary cultured cancer cells from patient tumors have been reported to maintain similarity with the original tumors with respect to histopathology, biomarker expression, genomic mutation profiles, and drug responsiveness [14]. Therefore, primary cultured cells have been suggested as a substitute preclinical model of various tumors for screening and evaluation of anti-cancer therapeutic candidates.
The traditional approach to drug discovery involves de novo identification and validation of new molecular entities, which is a time-consuming and costly process [15]. Despite huge investments in drug discovery and development, and explosive advances in biological technologies during the past decades, the number of new drugs introduced into the clinic has not increased accordingly. Alternatively, identification of new therapeutic indications for approved drugs i.e., drug repurposing has attracted particular attention for anti-cancer drug discovery [15-17]. This strategy has several advantages over the traditional de novo drug discovery approach, including reducing development time, costs, and the risks associated with pharmacokinetics and toxicology [17-20]. For drug repurposing research, several established clinical drug libraries approved by the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), and other agencies are available.
In the present study, we conducted drug repurposing study and could select guanabenz acetate (GA), an antihypertensive drug, as a candidate therapeutic molecule by high throughput screening on patient derived primary cultured HCC cells using the Prestwick Chemical Library which are all approved by the U.S. FDA and EMA. Mechanistically, GA inhibited growth arrest and DNA damage-inducible protein 34 (GADD34)-mediated dephosphorylation of eukaryotic initiation factor 2α (eIF2α), and subsequently induced apoptosis and autophagy of the HCC cells.
MATERIALS AND METHODS
Ethical approval
This study was approved by the Institutional Review Board of Asan Medical Center with a waiver of informed consent (IRB No. 2012-0112).
Patients and samples
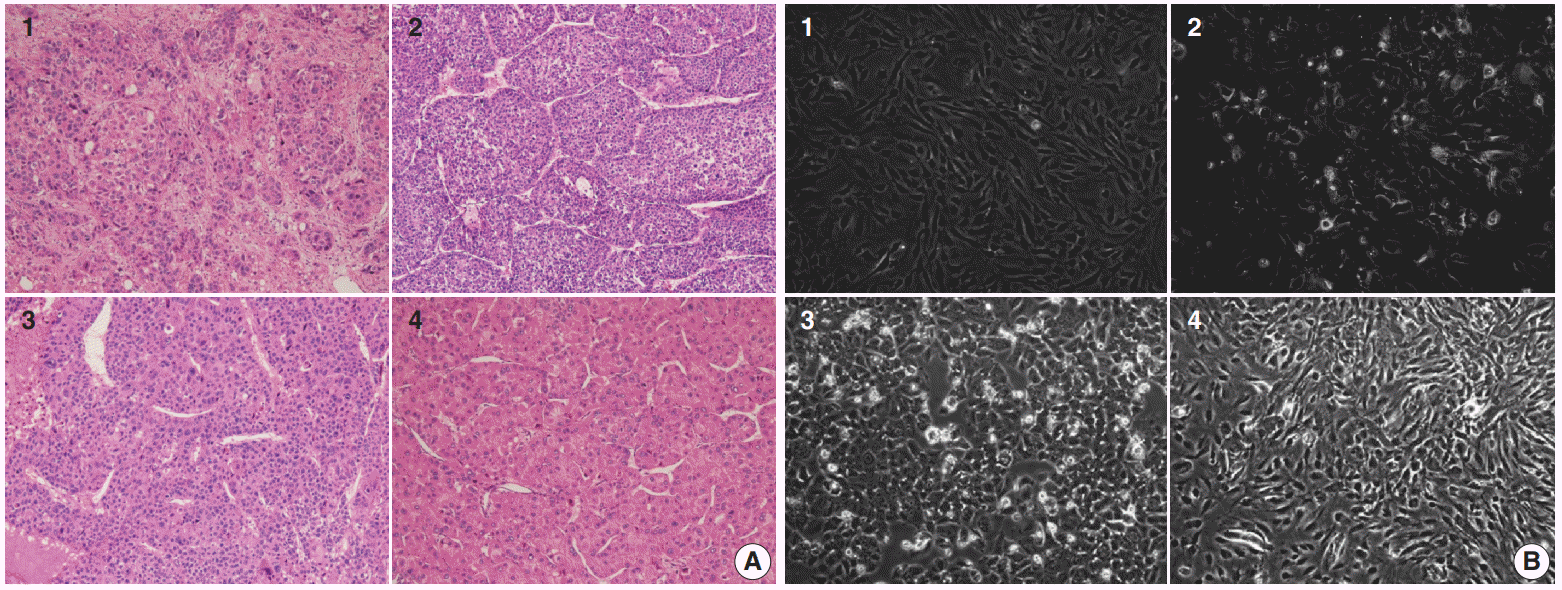
Four patients with HCC, who were confirmed to have the disease via liver protocol dynamic computed tomography scans at Asan Medical Center, Seoul, Korea, were selected to establish primary cultured cells. A portion of the tumor tissues obtained from surgical resection were fixed in cold 2% formaldehyde for 4 hours and embedded in paraffin at 56°C. Sections from the paraffin blocks (4-μm thick) were stained with hematoxylin and eosin (H&E). All H&E slides were reviewed by two pathologists (E.Y. and H.J.K.) who were blinded to the clinical information.
Primary culture of HCC cells
The fresh tissues of the four cases obtained during surgery were put into a tube containing Dulbecco’s modified Eagle medium: nutrient mixture F-12 (DMEM/F12; Sigma-Aldrich, St. Louis, MO, USA), with penicillin and streptomycin, and were transported to the tissue culture room. After removing normal liver tissue and connective tissue, tumor tissues were minced with scissors and subsequently digested with 0.1% type IV collagenase (Sigma-Aldrich) in a shaking incubator for 60 minutes at 37°C. After incubation, tissues were washed three to four times with DMEM/F12 containing 10% fetal bovine serum (FBS; Sigma-Aldrich). The HCC pellets obtained after centrifugation were plated on collagen type I dishes and incubated at 37°C in a 5% CO2 atmosphere. To favor the adhesion and growth of epithelial tumor cells, hepatocyte basal media containing human epidermal growth factor, transferrin, hydrocortisone, bovine serum albumin, ascorbic acid, GA-1000 (gentamicin and amphotericin B), insulin, 10% FBS, and a Triiodothyronine-SingleQuots kit (Biocompare, South San Francisco, CA, USA) were used to culture primary HCC tumor cells. The cultured HCC cells were harvested and stored in liquid nitrogen.
MTT cell proliferation assay/drug sensitivity assay
Two hundred and fifty-two small molecules from the Prestwick Chemical Library (Prestwick Chemical, Illkirch, France) were blindly selected for screening of anti-cancer effect. The primary cultured HCC cells (103 cells/well) were seeded in collagen type I coated 96-well plates. Following a 24-hour incubation at 37°C, the cells were treated with 252 small molecules (20 μM each). After 72 hours, 50 mg/mL of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reagent were added to each well, and the plates were incubated for 4 hours at 37°C. After the reduced formazan precipitate was dissolved in dimethyl sulfoxide, the absorbance at 540 nm was measured using a microplate reader (EL800, BioTek, Winooski, VT, USA). The concentration of candidate molecules to inhibit 50% of cell viability (IC50 value) was calculated using CalcuSyn Software (Biosoft, Cambridge, UK). These results were validated in six stable HCC cell lines (i.e., SNU398, SNU423, SNU449, SNU475, Hep3B, and Huh7). Five HCC cell lines (i.e., SNU398, SNU423, SNU 449, SNU475, and Huh7) and one HCC cell line (Hep3B) were respectively grown in RPMI 1640 medium (Sigma-Aldrich) and Minimum Essential Medium (Sigma-Aldrich), both supplemented with 10% FBS, penicillin, and streptomycin.
Small interfering RNA transfection
Hep3B and Huh7 HCC cells were treated with small interfering RNA (siRNA) specific to GADD34 and non-specific siRNA (Genolution Pharmaceuticals Inc., Seoul, Korea). Targeting sequences of siRNA were as follows: siGADD34-1, 5'-GGAUAAGGAAGAUGAUUCAUU-3'; siGADD34-2, 5'-GCCUAUAAUUUAUUAACUAUU-3'; and as a negative control, non-specific siRNA, 5'-CCUCGUGCCGUUCCAUCAGGUAGUU-3'. HCC cells were transiently transfected with GADD34 siRNA or non-specific siRNA with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Six hours later, the medium was replaced with regular culture medium, and the cells were incubated. The efficiency of silencing was assessed via western blotting 48 hours after transfection.
Western blot analysis
Harvested cells were lysed in Cell Lysis Buffer (Cell Signaling Technology, Beverly, MA, USA) for 20 minutes and centrifuged at 13,500 rpm for 5 minutes. Twenty micrograms of proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using 8%–12% gels and transferred to nitrocellulose membranes (Potran nitrocellulose membrane, Whatman, Kent, UK) using an iBlot dry blotting system (Invitrogen). The membranes were blocked with 5% non-fat dried milk and incubated with specific primary antibodies for (1) GADD34 (1:1,000), activating transcription factor 4 (ATF4; 1:1 000), p53 (1:1,000), and β-actin (1:2,000) from Santa Cruz Biotechnology (Dallas, TX, USA); (2) eIF2 (1:1,000), phospho-eIF2 (p-eIF2, 1:1,000), p-p53 (Ser15, 1:2,000), p21Waf1/Cip1 (1:500), cyclin B1 (1:1,000), cyclin E2 (1:1,000), p-cdc2 (p-cdk1, 1:2,000), Bax (1:1,000), Bcl-xL (1:2,000), procaspase-9 (1:2,000), and cleaved caspase-9 (1:1,000) from Cell Signaling Technology. After incubation with appropriate horseradish peroxidase-conjugated secondary IgG antibodies, bands were detected using ECL reagent (Amersham-GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Cell cycle assay
The effect of the agent on the cell cycle of HCC cell lines was analyzed by flow cytometry using propidium iodide (PI) staining. Briefly, 106 cells were seeded in 60-mm dishes and incubated with the agent (30 μM) or phosphate-buffered saline (PBS) as a control for 24 hours at 37°C. After incubation, the cells were fixed with ice-cold 70% ethanol at 4°C overnight, washed twice with PBS, incubated with 10 μg/mL RNase A, and stained with 30 μg/mL PI. The stained cells were subsequently analyzed using flow cytometry (FACSCalibur machine, Becton Dickinson, Mountain View, CA, USA) with a 560-nm dichroic mirror and a 600-nm pass filter (bandwidth, 35 μm). The percentages of cells in G1, S, and G2 phases were determined using Cell Quest 3.1 software (Becton Dickinson).
RESULTS
GA screened as a potential therapeutic drug for HCC
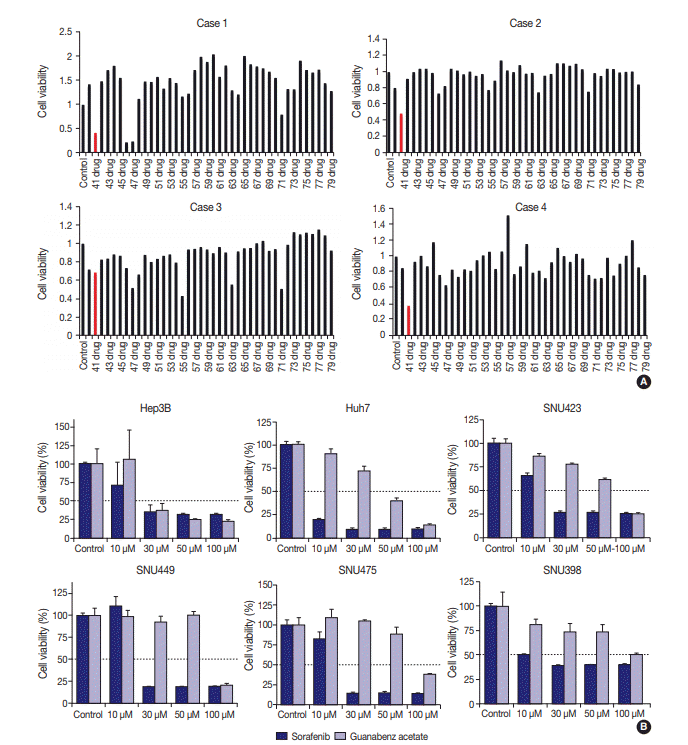
Four primary cultured HCC cells were established using surgically resected fresh tumor tissues from four HCC patients and subsequently confirmed via pathologic examination (Fig. 1A, B). The clinicopathologic features of the patients are listed in Table 1. High-throughput drug screening using the Prestwick Chemical Library, which contains 1,120 FDA-approved drugs (Supplementary Table S1), was performed on the four primary cultured HCC cells. Two hundred and fifty-two of the 1,120 molecules were randomly selected and screened and identified GA as a candidate. The results were confirmed using individual in vitro assay. After 72-hour incubation with a concentration range of the molecules (0–50 μM), cell viability was inhibited more than 50% with 20 μM GA (i.e., compound 41) (Fig. 2A) in three out of four primary cultured HCC cells, measured using the MTT assay.
GA inhibited growth of HCC cell lines
To further validate the anti-cancer effect of GA in HCC, six HCC cell lines were treated with GA (Sigma-Aldrich) or with a control molecule, sorafenib. After 72-hour incubation with GA, growth of Hep3B (IC50 = 30 μM) and Huh7 (IC50 = 50 μM) were inhibited (Fig. 2B). The IC50 values of GA for SNU423, SNU449, and SNU475 cells were 100 μM, and the viability of SNU 398 cells did not drop below 50%, even at 100 μM GA (Fig. 2B). When the inhibitory effects of sorafenib on cell proliferation were compared with those of GA, the decline in viability occurred at similar or lower concentrations with sorafenib in all six HCC cell lines (Fig. 2B).
GA targets PERK signaling pathway through GADD34 inactivation
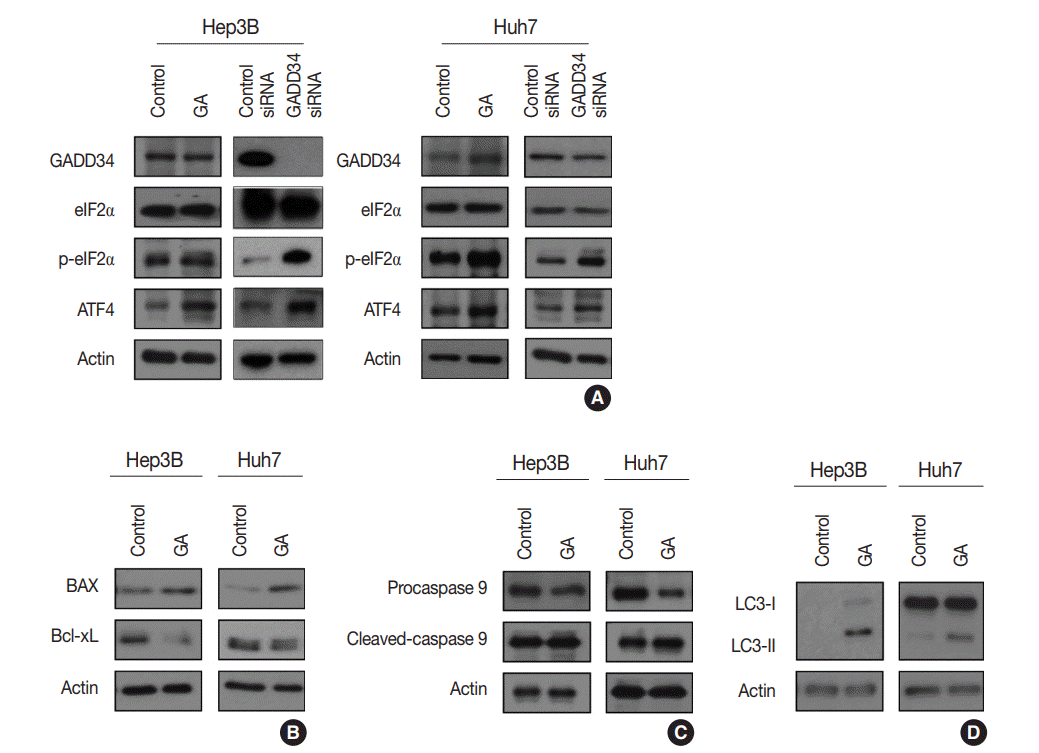
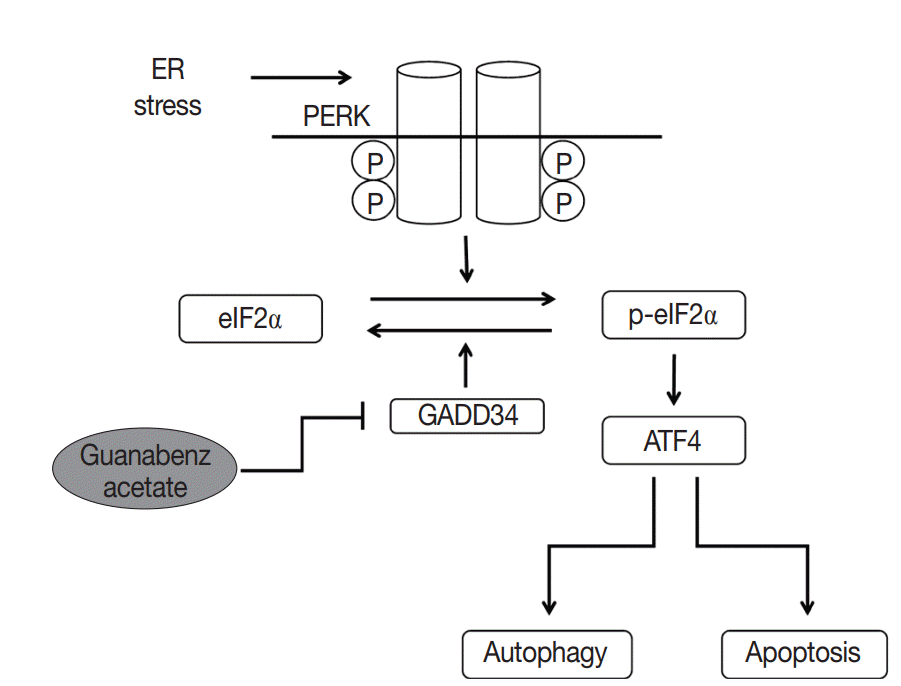
GA is an agonist of the α2 adrenergic receptor that is used as an antihypertensive drug. GA is also known to block dephosphorylation of eIF2α by inactivation of GADD34 in the protein kinase RNA-like ER kinase (PERK) signaling pathway, which would finally lead to expression of genes involved in apoptosis and autophagy. Therefore, the protein expression levels of genes associated with PERK signaling were evaluated by western blotting in the Hep3B and Huh7 cell lines to study the mechanisms of GA in HCC. The level of p-eIF2α was upregulated, and ATF4 protein was increased after GA treatment of Hep3B and Huh7 cells (Fig. 3A). In addition, when HCC cells were transfected with GADD34 siRNA, levels of p-eIF2α and ATF4 were upregulated in both Hep3B and Huh7 HCC cells compared with levels in control HCC cells (Fig. 3A). These results reveal that GA acts as an inhibitor of dephosphorylation of eIF2α, likely by inactivation of GADD34 in HCC.
GA induces apoptosis and autophagy-related cell death in HCC cells
ATF4, a transcription factor that induces the expression of genes, is involved in amino acid responses, metabolism, antioxidant responses, apoptosis, autophagy, and GADD34-mediated effects. To assess if GA induces HCC cell death via apoptosis and/or autophagy following ATF4 upregulation, expression levels of apoptosis-related proteins (i.e., Bax, Bcl-xL, procaspase-9, and cleaved caspase-9) and an autophagy-related protein (i.e., microtubule-associated protein light chain 3, LC3) were examined in Hep3B and Huh7 by western blotting. In the Hep3B and Huh7 cells treated with GA, Bax protein expression was higher, while Bcl-xL protein expression was lower than the expressions in control HCC cells (Fig. 3B). In addition, protein levels of procaspase-9 were decreased, while cleaved caspase-9 was increased in both Hep3B and Huh7 cells (Fig. 3C). Both LC3-I and LC3-II protein levels increased in Hep3B, but only LC3-II protein level increased in Huh7 treated with GA (Fig. 3D). These results clearly demonstrate that both mechanism of apoptosis and autophagy play a role in GA-induced cell death. Activated ATF4 in the PERK pathway is considered an essential gateway for apoptosis and autophagy in HCC cells.
Different types of cell cycle arrest in different HCC cell lines treated with GA
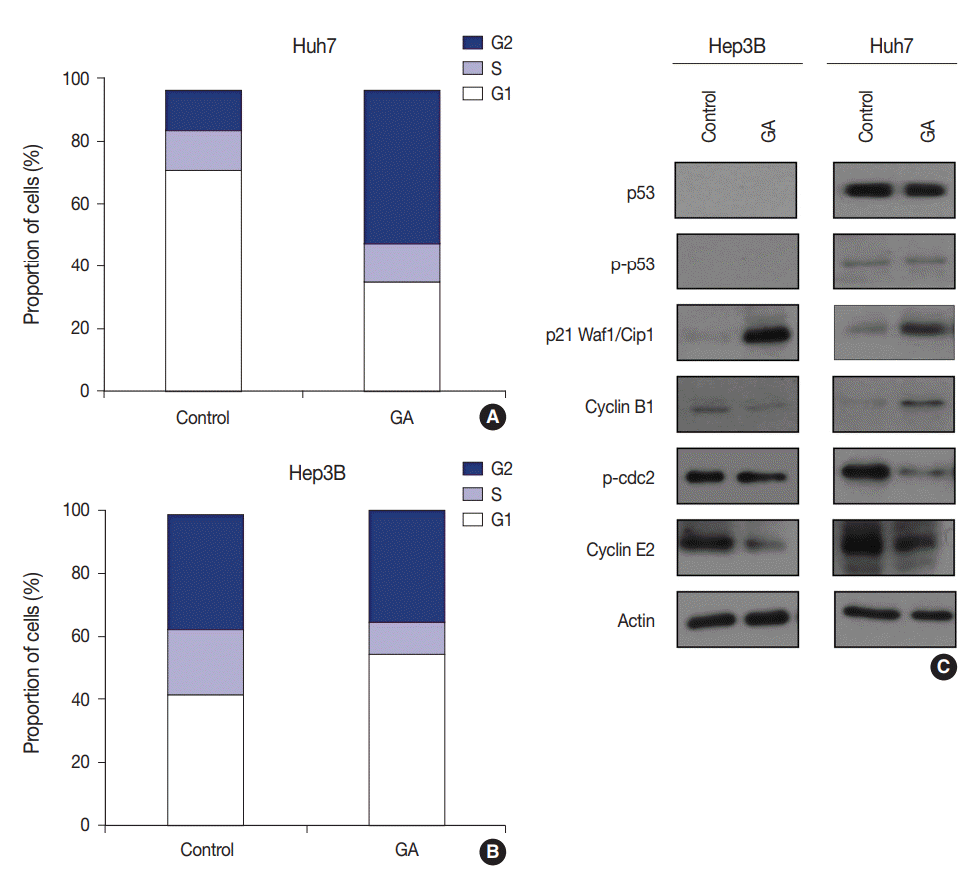
To examine whether GA reduces HCC cell viability by inducing cell cycle arrest, flow cytometry was performed. The expression levels of checkpoint proteins in the G1/S and G2/M transitions and cell cycle regulating factors were then examined by western blotting. The cell cycle distribution pattern was different between the two HCC cell lines. In Hep3B cells treated with GA, the percentage of G1 phase cells increased, while the percentages of S and G2 phase cells concomitantly decreased (Fig. 4A). In contrast to Hep3B, the cell cycle distribution of Huh7 treated with GA revealed an increase in the percentage of G2 phase cells, while producing a concomitant decrease in the percentage of G1 phase cells (Fig. 4B). Among cell cycle-related proteins, the total protein level of p21 increased remarkably after GA treatment in Hep3B and Huh7 cells. The protein level of cyclin B1 slightly decreased, p-cdc2 remained unchanged, and cyclin E2 decreased in Hep3B cells. In Huh7 cells, however, the protein level of cyclin B1 increased, p-cdc2 remarkably decreased, and cyclin E2 slightly decreased (Fig. 4C). These results suggest that the reduced viability of HCC cells by GA treatment is regulated by cell cycle arrest. Moreover, the patterns of cell cycle arrest by GA treatment can vary among different HCC cell lines.
DISCUSSION
In this study, we discovered that GA can be repurposed as a potential anti-cancer drug for HCC patients via preclinical drug efficacy screening by using primary cultured HCC cells that we established using the surgically resected HCCs and stable HCC cell lines.
A new functional mechanism of GA was recently reported. In this mechanism, PERK activity, which is one of three pathways of the unfolded protein response (UPR) in the endoplasmic reticulum (ER), is increased by selective inactivation of GADD34-mediated dephosphorylation of eIF2α, subsequently inducing apoptosis and autophagy (summarized in Fig. 5) [21-24]. UPR acts as a protective mechanism to alleviate ER stress by increasing protein-folding capacity, inhibiting general protein translation, and promoting degradation of unfolded or misfolded proteins. Interestingly, the aggregation of unfolded or misfolded proteins resulting in ER stress is associated with progression of various diseases, such as cancer, metabolic diseases, diabetes, inflammation, liver dysfunction, neurodegenerative disorders, and brain and heart ischemia [22-25]. Moreover, when ER stress is persistent and cannot be resolved, apoptotic signaling is initiated, which eventually leads to cell death [23]. Thus, the UPR signaling pathway can be an attractive target for drug discovery in diverse diseases. Recent studies have demonstrated that GA acts as a novel therapeutic agent by inhibition of dephosphorylation of eIF2α in the UPR pathway for several disorders, including progressive neurodegenerative disease (especially amyotrophic lateral sclerosis), toxoplasmosis, and breast cancer [21,26-28].
In the cases of HCC that were responsive to GA, the mechanisms of action were consistent with the known mechanism. First, we confirmed that GA acts on the PERK signaling pathway possibly by inactivation of GADD34 in HCC. The HCC cells treated with GA showed an increase in the phosphorylation of eIF2α and expression of ATF4. Moreover, both GA-treated HCC cells and GADD34 siRNA-transfected HCC cells showed the same patterns of p-eIF2α and ATF4, supporting that GA inhibits GADD34. Second, we also identified that GA reduced cell viability by both mechanisms of apoptosis and autophagy in HCC cells. In GA-treated HCC cells, apoptosis and autophagy reactions were confirmed by decreased expression of procaspase-9 and the increased levels of cleaved caspase-9 and LC3. We also discovered that GA independently affects the cell cycle. GA-treated HCC cells showed arrest in the G1 or G2 phase of the cell cycle, the pattern of which was different between the two HCC cell lines. Additional studies are needed to elucidate the reason why the phase of cell cycle arrest is different among HCC cell lines, and the mechanisms of these differences. In addition, the markedly upregulated p21Waf1/Cip1 protein levels in both Hep3B and Huh7 cells indicate that a p53-dependent pathway could be involved in the cell cycle arrest by GA.
The present study is the first to perform a drug repurposing study using primary cultured cells that we established from HCC patients with a high-throughput screening approach to discover novel therapeutics. The drug discovery process was developed based on genome-based drug discovery, high-throughput screening, and combinatorial chemistry [17]. Contrary to expectations, novel agents identified through traditional methods have gradually declined because of high costs, time-consuming processes, and unexpected adverse reactions in clinical trials. This has led to a process called drug repositioning or repurposing [29,30]. Drug repositioning is a new approach for drug discovery and is believed to offer great benefits over the traditional method of searching for new active substances, since the safety and pharmacokinetics have already been established [17,18,31,32]. A representative new indication, introduced by drug repositioning, is thalidomide, a sedative hypnotic agent, which demonstrated significant clinical activity against multiple myeloma and leprosy [33,34]. Moreover, the combination therapy of drugs found by drug repositioning and conventional anticancer drugs may increase efficacy and reduce adverse reactions [17]. Thus, drug repositioning may become a key approach for treating variable malignancies, including HCC, in novel drug discovery.
The commercially available libraries of marketed drugs and natural compounds have been widely used in drug screening tests for a broad spectrum of diseases, including cancers, viral or fungal infections, neurodegenerative disorders, and neuromuscular disorders, but never have been used for drug efficacy tests of HCC [35]. Although the unexpected effects of GA on the reduction of HCC cell viability were discovered from randomly selected drugs, the effects of GA on malignant cells have already been reported in breast cancer. A previous study revealed that combined treatment of GA and salubrinal can attenuate the malignant phenotype and tumor growth of triple negative breast cancer cells through the eIF2α-mediated Rac1 pathway [28]. As mentioned previously, GA acts as an inhibitor of dephosphorylation of eIF2α and can be used potentially as an anti-cancer drug for other malignancies, especially those originating from ER-rich organs.
This study has a few limitations. It was performed in vitro and the GA IC50 value is high for a clinically achievable dose. Therefore, additional in vitro and in vivo studies are needed to conclusively determine whether GA can be a novel anti-cancer agent for inoperable HCC patients at a clinically achievable dose. In addition, molecular and immunohistochemical studies are needed to determine the predictive marker(s) for the therapeutic effects of GA in HCC patients. Finally, the effect of GA as an anti-cancer agent could be evaluated in other malignancies of ER-rich organs.
In conclusion, GA, which was screened in primary cultured HCC cells, reduces HCC cell viability via the combined effects of apoptosis, autophagy, and cell cycle arrest. Therefore, GA may be repositioned as an anti-cancer therapeutic agent for HCC patients.
 XML Download
XML Download