

 PDF
PDF Citation
Citation Print
Print



Beckwith-Wiedemann syndrome (BWS, OMIM 130650) [1]. characterized by hemihypertrophy, macroglossia, macrosomia, organomegaly, hyperinsulinism, and distinct facial features [2]. The genetic causes of BWS are complex but are mainly due to genomic imprinting disorders involving chromosome 11. In addition, patients with BWS are predisposed to developing tumors, with the incidence of the tumor being 4% to 21%. Most tumors consist of embryonal tumors such as Wilms tumor, hepatoblastoma, neuroblastoma, adrenal cortical carcinoma, or rhabdomyosarcoma [3,4]. Given this disposition, a thorough tumor screening is recommended for BWS patients. We recently experienced a BWS patient who had adrenal cortical neoplasm with uncertain malignant potential arising in the heterotopic adrenal cortex located in his liver.
CASE REPORT
A male infant who was the third child of healthy parents was born at 36 weeks of gestation weighing 4,150 g. After birth, he was hospitalized in a neonatal intensive care unit for 10 days due to cyanosis and neonatal jaundice. Due to the presence of right hemihypertrophy and macroglossia, he was suspected of having BWS, and a genetic test for BWS confirmed paternal uniparental disomy (KvDMR/H19 aberrant methylation T0392241) in chromosome 11. At the time of birth, no mass was found in the imaging work-up.
At nine months, the infant’s weight was in the 97th percentile at 11.7 kg, and his height was in the 95th percentile at 77 cm. Exercise development was average; however, due to leg length discrepancy by the prominent right hemihypertrophy, the infant required an aid to move. There was no significant virilization.
During the tumor screening, abdominal ultrasonography revealed a 1.9 × 1.2 cm oval-shaped hypoechoic mass in the inferior tip of his right liver (Fig. 1A). A computed tomography (CT) scan showed an exophytic mass arising from the inferior tip of his right lobe of the liver (Fig. 1B). The mass had heterogenous enhancement on the portal venous phase of contrast-enhanced abdominal CT scan. Apart from this, there were no abnormal findings in the kidneys, adrenal glands, lymph nodes, or other abdominal organs. Based on the ultrasound and CT imaging, hepatoblastoma of the liver was suspected. The levels of aspartate aminotransferase and alanine aminotransferase were in the normal range at 34 IU/L and 30 IU/L, respectively. Serology testing for hepatitis was negative. The level of α-fetoprotein was slightly elevated at 57.4 ng/mL.
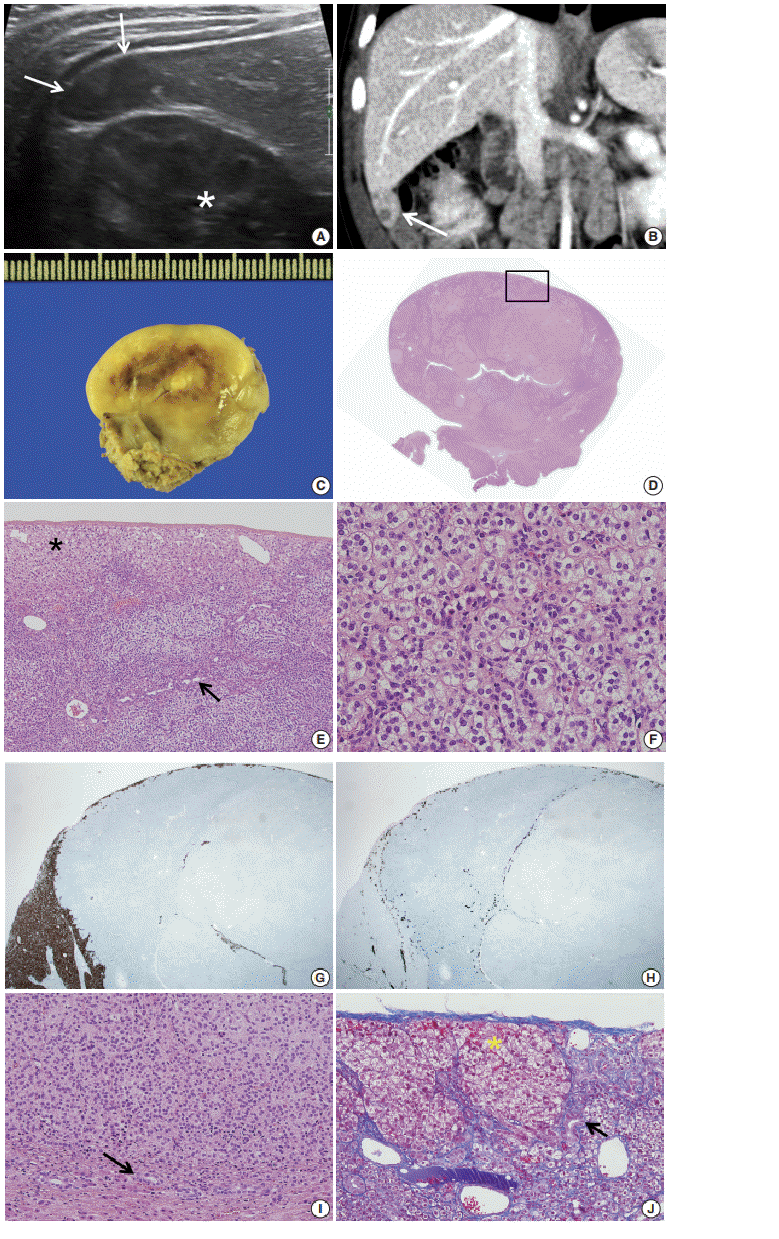 | Fig. 1.(A) Axial ultrasound image showing an oval-shaped hypoechoic mass in the inferior tip of the right hemi liver (arrows). Asterisk indicates the right kidney. (B) Coronal computed tomography scan showing an exophytic mass arising from the inferior tip of the right hemi liver (arrow). (C) The cut surface of the tumor is grayish yellow with focal brown pigmentation and glistening. Necrosis and hemorrhage are absent. (D) The tumor shows exophytic growth (scan view). (E) The tumor is encased in a Glisson capsule and had remaining hepatocytes (asterisk). Bile ductules (arrow) were entrapped. (F) Tumor cells with nested growth pattern had plump clear cytoplasm, distinct and smooth cell membrane, bland and small nucleus, and micro nucleoli that resemble zona fasciculata of a normal adrenal gland. (G) Hepar-1 immunohistochemical staining showing the remaining normal hepatocytes surrounding tumor cells. (H) Cytokeratin 7 immunohistochemical staining showing the entrapped bile ductules in the periphery of tumor cells. (I) The bile ducts (arrow) are not in contact with the tumor. (J) Remaining bile ducts (arrow) and hepatic parenchyma (asterisk) are pushed by the tumor. The Glisson capsule remains intact (Masson trichrome staining).
|
Suspecting hepatoblastoma, a laparoscopic partial hepatectomy was performed. Grossly, the resected tumor was accurately demarcated, revealing a solid mass measuring 2.5 × 1.5 × 0.6 cm. The cut surface of the tumor was grayish white, yellow, and glistening (Fig. 1C). Microscopically, the tumor showed a nested growth pattern (Fig. 1D). The tumor cells had plump clear cytoplasm, distinct and smooth cell membranes, bland and small nuclei, and inconspicuous nucleoli which resembled the zona fasciculata of a normal adrenal gland (Fig. 1E, F). Mitosis was found in two out of 10 high-power fields, and there was no atypical mitosis. A Glisson capsule encased the tumor. Normal hepatic parenchyma and the remaining bile ducts were highlighted by Hepar-1 and cytokeratin 7 (Fig. 1G–J), and the remaining liver parenchyma was unremarkable (Fig. 1G–J).
We performed immunohistochemical staining using formalin-fixed, paraffin-embedded, 4-μm-thick tissue sections with an OptiView DAB immunohistochemical detection kit.
The tumor cells did not exhibit immunoreactivity for arginase-1, hepatocyte (Hep par 1), α-1-fetoprotein, glypican 3, β-catenin, or glutamine synthetase. INI-1 was expressed in the nucleus. The results showed that this was not a case of hepatoblastoma.
To confirm that the tumor originated from adrenal glands, immunohistochemistry for CD56, vimentin, Melan A, inhibin α, synaptophysin, and chromogranin was performed. The tumor cells showed strong diffuse immunoreactivity for CD56, vimentin, moderately intense immunoreactivity for Melan A, and weak and focal immunoreactivity for inhibin α. Synaptophysin immunoreactivity was weak and focal. The result of chromogranin immunohistochemistry was negative, and Ki-67 labeling index was 13.2% by manual count (Fig. 2A–F). Detailed information and results of the immunohistochemistry are described in Table 1. Reticulin histochemical staining was performed to evaluate the reticulin framework. As a result, reticulin staining demonstrated multifocal disruption of reticulin network within tumor cells (Fig. 2E, F), indicating that this tumor has uncertain malignant potential.
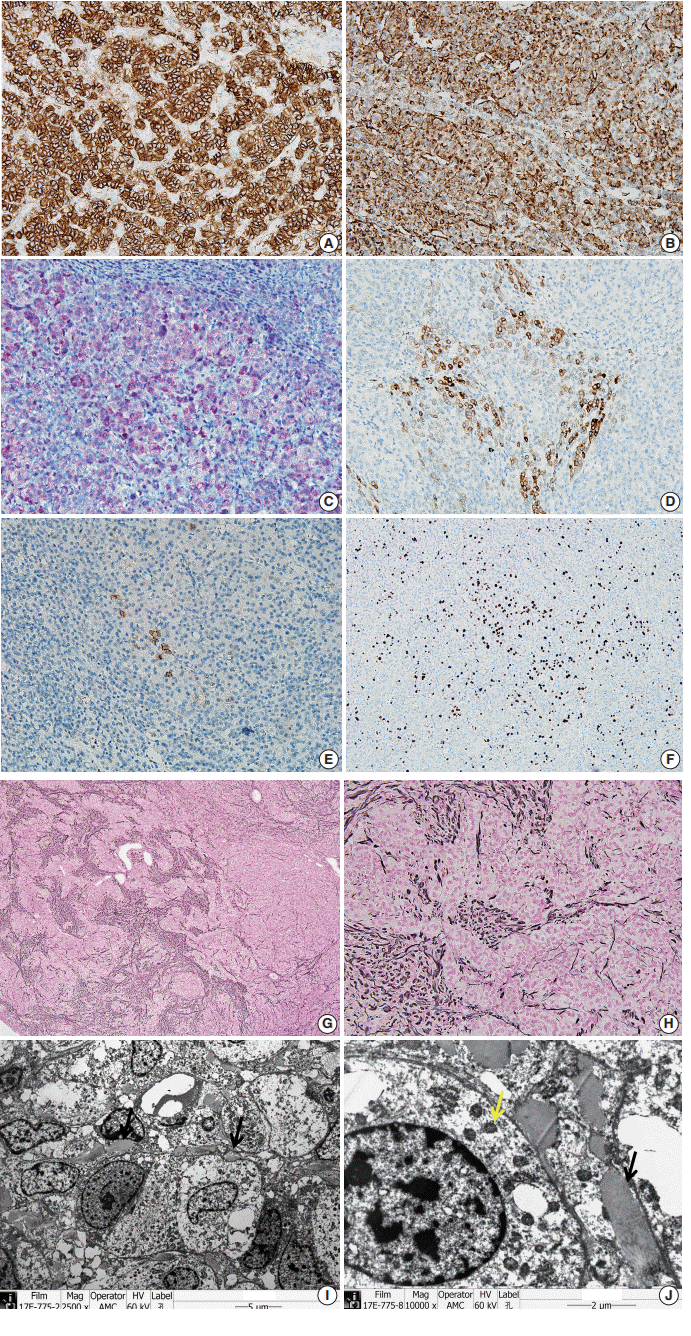 | Fig. 2.The tumor cells show strong, diffuse immunoreactivity for CD56 (A), Vimentin (B), moderately intense immunoreactivity for Melan A (C), and weak focal immunoreactivity for inhibin α (D). (E) Synaptophysin immunoreactivity is very focal and weak. (F) Ki-67 labeling index is 13.2% in hot spot by manual count. (G, H) The tumor shows multifocal disruption of reticulin network. Electron microscopy shows numerous mitochondria with tubular cristae (I, ×2,500; J, ×10,000, black arrow) and neurosecretory granules (J, yellow arrow), which are compatible with adrenal cortical neoplasm.
|
Table 1.
Detailed information and results of immunohistochemistry
![]()
Electron microscope
Ultra-thin sections from formalin fixed tissue were examined with a transmission electron microscope (model GEM-1400, Jeol, Tokyo, Japan). Electron microscopy revealed numerous mitochondria with tubular cristae and neurosecretory granules, which are compatible with the characteristics of adrenal cortical neoplasm (Fig. 2I, J). Histologic findings, immunohistochemical results, and electron microscopic findings all strongly suggested the possibility of adrenal cortical tumor present in the liver. The bilateral adrenal gland was normal, and there was no evidence of a primary tumor in the adrenal gland; therefore, we concluded that the origin of the adrenal tissue was of heterotopic nature.
Since only the pathologic findings of the tumor and only deidentifed personal information of the patient were used, formal written informed consent was not required with a waiver by the appropriate IRB of Asan Medical Center (IRB number: 2018-1042).
Go to : 
DISCUSSION
The heterotopic adrenal tissue is rarely found in the lungs, brain, ovaries, or placenta [5]. Heterotopic adrenal tissue tends to occur in the abdominal cavity along the gonadal descent pathway or celiac axis [6]. There are a few reports of heterotopic adrenal cortex in the liver [7,8].
There are two possible explanations for the mechanism of heterotopic adrenal cortex in liver parenchyma [8]. The first theory is a migration of adrenal gland tissue to the liver parenchyma along the mesogastrium during early developmental stage. The second theory is an occurrence of adreno-hepatic fusion during the later stage. During the fusion, the mesenchyme encases the adrenal gland and liver, and after the mesenchyme is separated, adrenal gland fragments are entrapped in the liver parenchyma [2,9]. Adreno-hepatic fusion is a relatively common phenomenon found in 9.9% of autopsy specimens [10], and is considered to be a part of the aging process as adreno-hepatic fusion is more often observed in older people. However, tumorous conditions arising from adreno-hepatic fusion are not common, and there is a partial capsule that separates the two organs.
In our current case, the tumor was encased by a Glisson capsule and hepatocytes and bile ductules found in the periphery of the tumor. And the patient was an infant. These characteristics suggest that the first theory discussed above regarding migration of adrenal gland tissue is likely the cause of the presence of heterotopic adrenal cortex tin the liver of this patient.
For patients with BWS, many embryonal tumors occur due to the effects of growth factors and cyclin-dependent kinase. Although rare, various adrenal gland tumorous conditions (including cytomegaly of the fetal adrenal cortex, benign cyst, hyperplasia, adenoma, adenocarcinoma of the cortex, neuroblastoma, and pheochromocytoma) can develop in BWS patients. There were reports of neoplasms from the heterotopic adrenal cortex located in the liver [7], hilum of the kidney [11] and the spinal cord [12] of BWS patients (Table 2).
Table 2.
Neoplasm arising in ectopic adrenal tissue in BWS: clinical and pathological features as described in the literature
| Reference | Age at diagnosis | Sex | Histologic diagnosis | Lesion location | Symptom | Management | Outcome | Associated disease |
|---|---|---|---|---|---|---|---|---|
| Wilkins et al. [7] | 2 yr 9 mo | M | Adrenocortical tumor | Liver | Virilism, Cushing syndrome | Right lobectomy | NED | No adrenal tissue in the usual location on the right |
| Cardinalli et al. [11] | 1 yr 5 mo | M | Myelolipoma | Renal hilum | Elevated blood level of cortisol, dehydroepiandrosterone | Adrenalectomy with ipsilateral renal hilar and intercaval-aortic lymph node dissection | NED | Adrenal cortical adenoma in adrenal gland |
| Giner et al. [12] | 2 yr | M | Adrenal cortical adenoma with oncocytic features | Spinal cord (L4-L5, extramedullary, intradural) | Abnormal gait | L3-S1 laminoplasty | 1 Year later recurrence at the same level → second surgery → NED |
![]()
The incidence of childhood adrenal tumor is minimal (0.5%), and it is challenging to distinguish malignant adrenal tumor from a benign one, especially in child patients. The tumor of this patient was small, and there was no evidence of metastasis, as evidenced by the fact that the bilateral adrenal glands were normal. The mitotic count was low, and neither necrosis, atypical mitosis, nuclear atypia nor lymphovascular invasion was observed. Only multifocal reticulin disruption was observed. Therefore, we concluded that the tumor was adrenal cortical neoplasm with uncertain malignant potential rather than adrenal cortical carcinoma or adrenal cortical adenoma, according to the diagnostic criteria of Wieneke, et al. [13]
Hepatoblastoma is the most common tumor in children with BWS. Therefore, it is possible that adrenal cortical neoplasm arising in the heterotopic adrenal cortex may be mistaken for hepatoblastoma in a usual clinical setting. To the best of our knowledge, our patient is the first case of adrenal cortical neoplasm with uncertain malignant potential arising from a heterotopic adrenal cortex observed in the hepatic parenchyma of a BWS patient. Pathologists will be able to avoid misdiagnosis if they know that tumors can occur from the heterotopic adrenal cortex in the liver especially in patients with BWS.
Go to :
 XML Download
XML Download