

 PDF
PDF Citation
Citation Print
Print



Adrenocortical carcinoma is a rare and heterogeneous malignancy with a poor prognosis, the pathogenesis of which is not yet completely understood. Patients present with hormone excess or local mass effect [1]. An adrenal rest (ectopic adrenal tissue) can occur anywhere along the gonadal descent. This tissue usually has no clinical significance, but it may become hyperplastic or malignant in patients with primary or secondary adrenal pathology [2]. Most adrenal rest tumors are functional and diagnosed preoperatively. However, the less frequent nonfunctional adrenal rest tumors are discovered accidentally or postoperatively [3]. We described a patient with a nonfunctioning intrarenal adrenocortical carcinoma that arose from an adrenal rest.
CASE REPORT
This study was approved by the Institutional Review Board of Severance Hospital with a waiver of informed consent (IRB No. 4-2017-1044).
A 61-year-old man was evaluated for back pain that had persisted for 10 days. Abdominopelvic computed tomography (APCT) and magnetic resonance imaging were performed at a local clinic and revealed a 13-cm mass in his right kidney. The mass was causing an ureteropelvic junction obstruction that broadly contacted the second and third duodenal portions, psoas muscle, and inferior vena cava. All radiologic findings were consistent with renal cell carcinoma with multiple lung metastases. The patient was hospitalized at our institution, and the APCT was repeated and provided the same diagnosis (Fig. 1A).
The patient underwent radical nephrectomy without adrenalectomy. The right kidney weighed 1,135 g and measured 17 × 12 × 6 cm. The tumor had a smooth and bulging external surface. Cross sections revealed a well-circumscribed and yellowish lobulated hard mass (Fig. 1B) measuring 14 × 12 × 8 cm, present in the mid pole of the right kidney. The mass showed extensive necrosis (60%) and hemorrhage (30%).
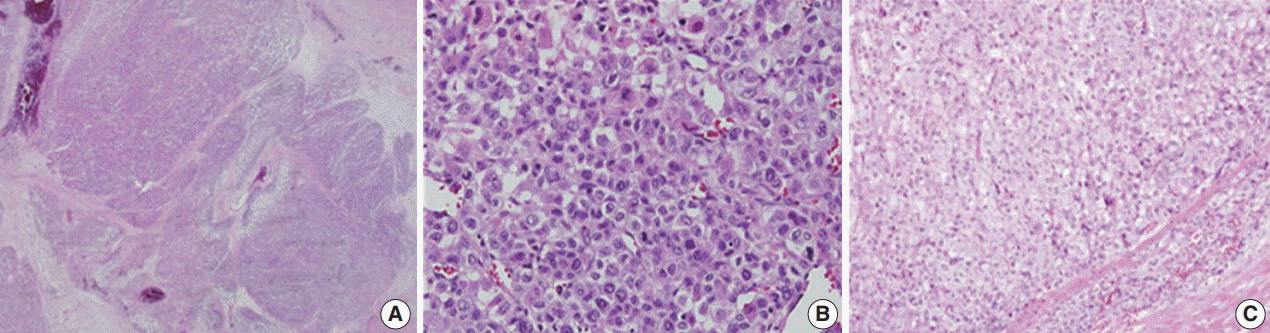
Microscopically, the tumor had multilobulated nests divided by thick fibrous septations (Fig. 2A). The tumor was comprised of compact polygonal cells with distinct cell borders and granular cytoplasm. Sinusoidal vascular ingrowth was less distinct. Nuclei were round or ovoid, hyperchromatic with central prominent nucleoli, and contained frequent mitoses (40/50 high-power fields) (Fig. 2B) without raisinoid nuclei or perinuclear haloes. Some areas containing adrenal cortical-like tissues were identified (Fig. 2C). Necrosis and vascular invasion were also present. These results suggested against the diagnosis of renal cell carcinoma.
DISCUSSION
Ectopic or accessory adrenal cells are often found postnatally along the path of gonadal descent because the adrenocortical primordium develops in close proximity to the urogenital ridge of the emerging gonad and migrates alongside the gonad [4]. Typically, these cells disappear within a few years of birth, and sometimes these cells linger without any event.
Ye et al. [4] reported seven cases of intrarenal adrenal tissue and two cases of renal-adrenal fusion. Except for one case identified within the kidney mid pole, all intrarenal lesions were found in the superior portion of the kidney. Our case reports a malignant tumor arising from the ectopic adrenal rest in the mid pole of the kidney. In all nine reported cases of Ye et al. [4], the intrarenal adrenal tissues were composed of only adrenal cortical tissue with no adrenal medullary tissue present. There is a recent review of the literature about adrenocortical carcinoma arising in an adrenal rest. Reported malignant tumors arising from an ectopic adrenal rest are predominantly adrenocortical carcinomas of the retroperitoneum, gonad, liver, kidney, spinal cord, and pelvis [5]. Intrarenal adrenocortical carcinomas have been previously identified in the hilum [3] and in the mid pole of the kidney as in our case. Adrenocortical carcinomas that involve the gonads show relatively high rates of mortality [6-8].
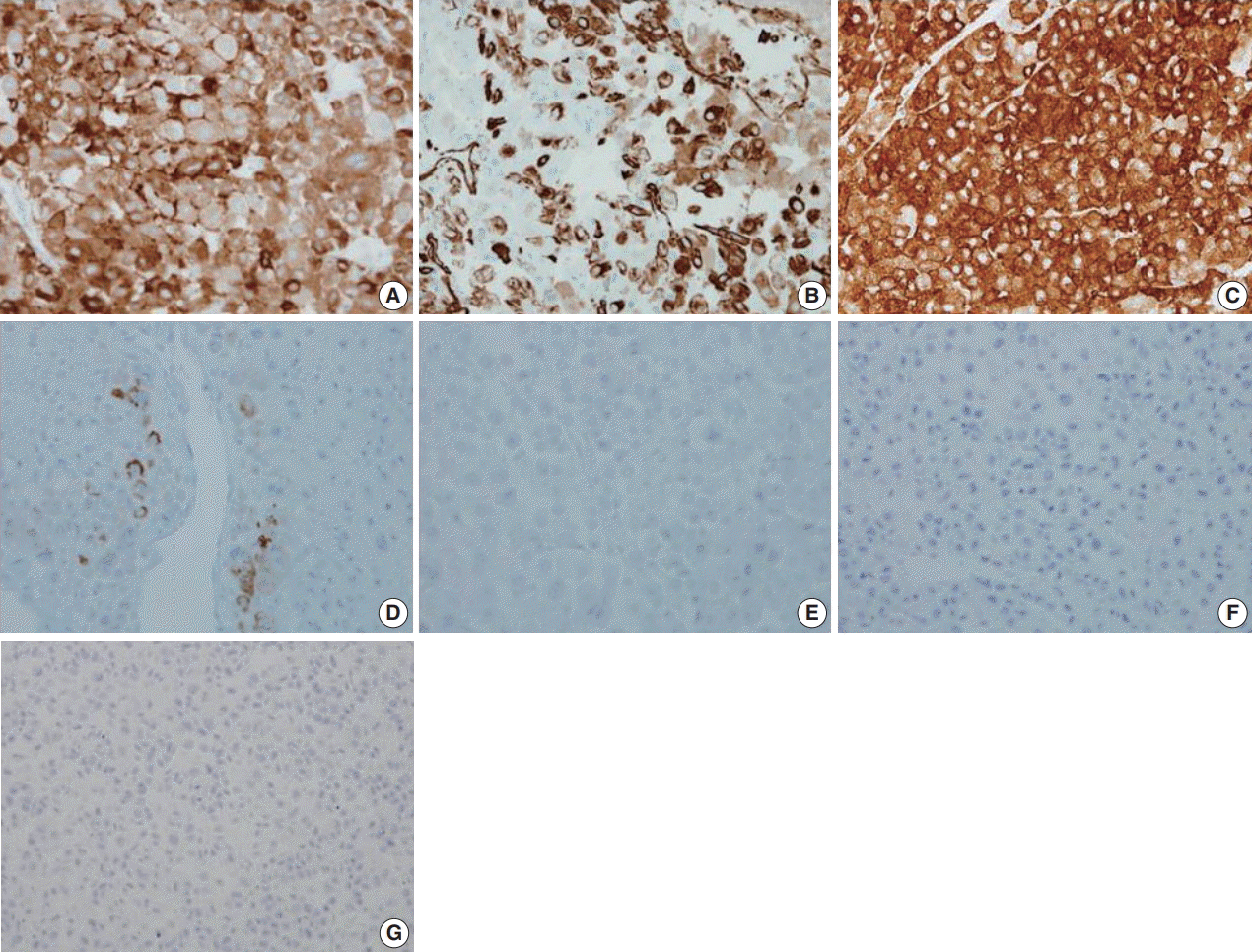
The microscopic features that favor the diagnosis of renal cell carcinoma over adrenocortical carcinoma are the presence of glands, particularly if they contain red blood cells, and abundant cytoplasmic glycogen. However, neither is pathognomonic and were present in the case. Among the nine histological parameters of the Weiss scoring system for histologic diagnosis of adrenocortical carcinoma (high nuclear grades [Fuhrman nuclear grades III and IV], mitotic rate > 5/50 high-power fields, atypical mitotic figures, clear tumor cell cytoplasm [less than 25% tumor cells], diffuse architecture [greater than 33% of tumor], necrosis, venous invasion, sinusoidal invasion, and capsular invasion), the present case met seven parameters, excluding sinusoidal invasion and capsular invasion. After the diagnosis of renal cell carcinoma was excluded by morphology and negative cytokeratin expression, we examined additional immunohistochemical stains to differentiate epithelioid angiomyolipoma, adrenocortical carcinoma, glomus tumor, or related mesenchymal tumors. Finally, we defined the tumor as adrenocortical carcinoma (pT2NxcM1).
Currently, radical surgery is the only curative approach, and it is recommended for all patients with resectable adrenocortical carcinoma tumors, including those patients with recurrent disease. There is no consensus concerning adjuvant therapy [9]. However, recent studies have reported that adjuvant mitotane may prolong recurrence-free survival in patients with radically resected adrenocortical carcinoma [10,11].
Our patient was treated with radical nephrectomy and adjuvant chemotherapy (VAP; vincristine, doxorubicin, and prednisolone) with mitotane. He has been healthy with no evidence of recurrence or metastasis for 3 months after the original diagnosis. Recent studies have reported the prevalence of adrenocortical carcinoma in Korea to be 2%–5%. In our institution, five cases of adrenocortical carcinoma were reported from 2000 to 2018. Among them, this is the only and first reported case of intrarenal adrenocortical carcinoma. We reported a rare case of intrarenal adrenocortical carcinoma arising from an ectopic adrenal rest as a mimicker of renal cell carcinoma in the kidney. Although the incidence of malignancy arising in an adrenal rest is low, clinicians and pathologists must be aware of the possibility because of its poor prognosis and common recurrence and metastasis.

 XML Download
XML Download