

 PDF
PDF Citation
Citation Print
Print


Rhabdomyosarcoma (RMS) is a malignant soft tissue tumor with a skeletal muscle phenotype that mainly affects children and adolescents [1]. Among its subtypes, alveolar rhabdomyosarcoma (ARMS) is more common in adolescents and young adults and has a worse prognosis, compared to embryonal rhabdomyosarcoma (ERMS) [2]. RMS frequently arises in the head and neck, genitourinary system and extremities and commonly metastasizes to lung and bone [1]. The central nervous system (CNS) can be involved by direct extension or distant metastasis, and CNS involvement may present either as a parenchymal lesion or leptomeningeal metastasis (LM) [3].
Although cerebrospinal fluid (CSF) cytology is considered as a gold standard for diagnosing LM, cytologic features of RMS in CSF has not been described to date, which makes it challenging for cytopathologists to recognize the involved CSF and further affects future therapeutic plans and prognosis. Here, we report a case of metastatic ARMS that was diagnosed by CSF cytology.
CASE REPORT
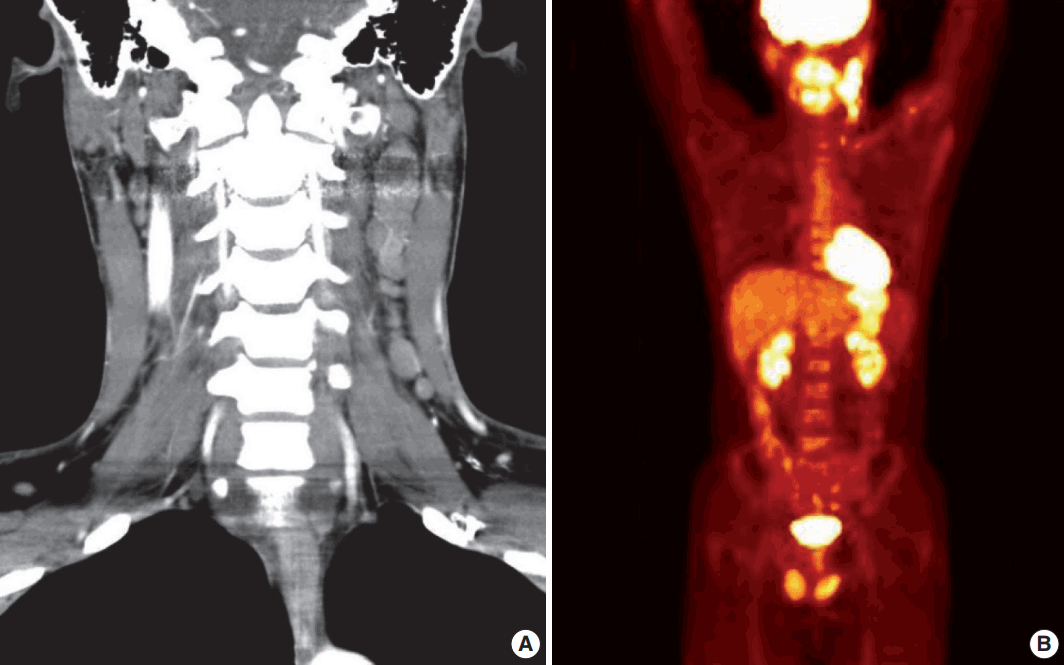
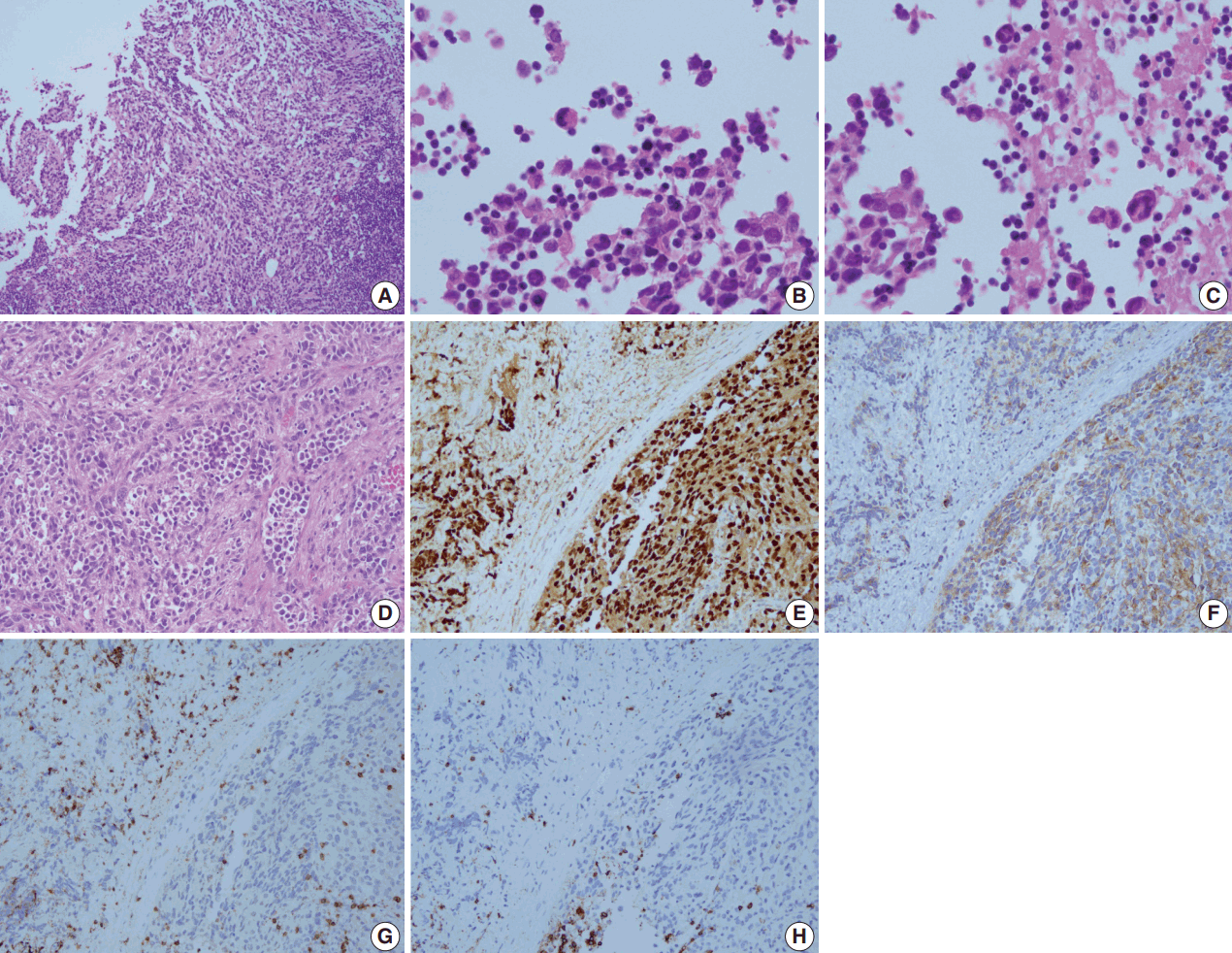
A 28-year-old man presented with a mass on the left side of the neck that had persisted for 4 months. On physical examination, a 3-cm firm, non-tender mass was palpated. Computed tomography showed multiple enlarged left cervical lymph nodes with homogeneous enhancement (Fig. 1A). An excisional biopsy of the cervical lymph node was performed. Microscopic examination revealed diffuse sheets of poorly differentiated cells with variable amounts of eosinophilic cytoplasm (Fig. 2A). A few cells showed abundant cytoplasm and eccentric nuclei, and multinucleated cells with peripherally located nuclei were observed (Fig. 2B, C). Immunohistochemistry (IHC) indicated that the tumor cells were negative for CD3, CD20, CD30, and cytokeratin and positive for vimentin and myogenin (Fig. 2E, G, H). The tumor was positive for anaplastic lymphoma kinase (ALK) (Fig. 2F). Diagnosis of metastatic ERMS was made, but the primary site of the tumor was not identified in positron emission tomography (Fig. 1B). Fluorescence in situ hybridization (FISH) did not reveal ALK gene rearrangement, however, a low-level gain of ALK gene copy number was observed.
The patient received two cycles of preoperative IA (ifosfamide, adriamycin) and neck dissection, followed by three cycles of VAC (vincristine, dactinomycin, cyclophosphamide) and five cycles of VC (vincristine, cyclophosphamide). He was in complete remission for 6 months before the tumor recurred in the bilateral neck and left retropharyngeal space. Biopsy of the recurrent lesion showed the same histology as the previous specimen, but was diagnosed as ARMS based on diffuse, strong immunoreactivity for myogenin. The tumor progressed despite further treatment with crizotinib and the patient subsequently received palliative IA, radiation therapy and VIP (etoposide, ifosfamide, cisplatin).
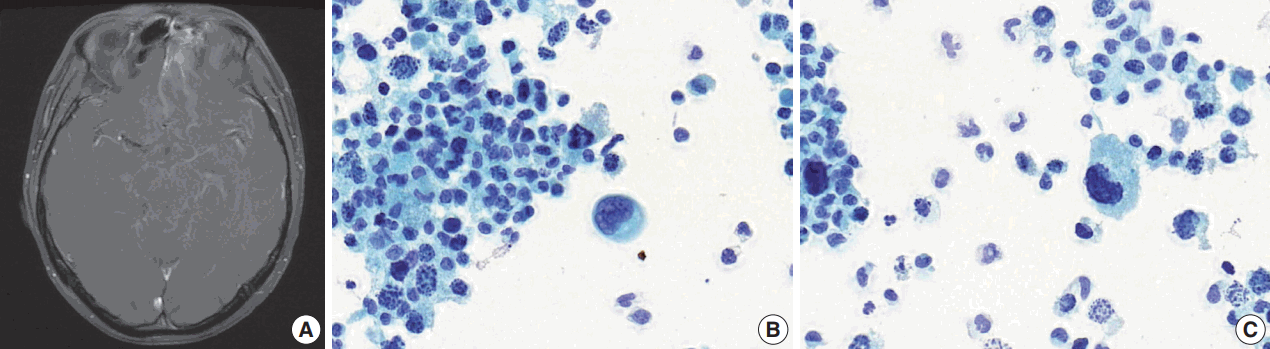
After 10 months of palliative therapy, he developed seizure-like movement, nausea, and vomiting. Brain magnetic resonance imaging demonstrated leptomeningeal enhancement of the bilateral rectus gyri, left orbital gyrus and brainstem, with enhancing soft tissue in the left ethmoid sinus (Fig. 3A). A spinal tap was performed and a Papanicolaou-stained preparation of CSF revealed atypical small-sized cells, mostly individually scattered, with some forming clusters (Fig. 3B). Large atypical cells with eccentric nuclei and abundant cytoplasm were identified. The tumor cells showed nuclear pleomorphism with occasional horseshoeshaped nuclei, and frequent karyorrhexis (Fig. 3C).
The CSF specimen was diagnosed as malignant tumor involvement. Intrathecal methotrexate was added but did not have an effect, and the patient refused further treatment.
This study was approved by the Institutional Review Board of the Seoul National University Hospital with a waiver of informed consent (IRB No. 1801-102-917).
DISCUSSION
LM usually occurs as a late-stage complication of malignancy, and due to development in systemic therapy and improved survival, the at-risk patient population has increased [4]. De et al. [3] reported 23 cases of RMS with CNS relapse, including 21 cases with LM. In their report, CNS relapse occurred from 1 to 23 months after initial diagnosis, with median of 12 months. LM results from dissemination of tumor cells in CSF, and tumor cells reach the CSF by hematogenous spread, direct extension or by growing along the blood vessel or nerve sheath [4]. In our case, the tumor was located in the cervical lymph node at initial diagnosis, but the metastatic lesion developed an intracranial extension and LM after going through multiple cycles of treatment and recurrence. Time from initial diagnosis to LM was 29 months.
To date, literature on cytologic feature of RMS has been mainly focused on fine-needle aspiration (FNA). There are few reports on body fluid cytology, and this is the first report on CSF. In previous studies on fluid cytology, RMS cells commonly appeared as single cells or as or clusters of small round cells with scant to moderate amounts of vacuolated cytoplasm [5]. Tumor cells show hyperchromatic and pleomorphic nuclei with occasional prominent nucleoli, and some authors have reported rhabdomyoblast-like, multinucleated or large pleomorphic cells with eccentric nuclei [6-8]. Cytoplasmic cross-striation is not recognized in most cases. In immunocytochemistry, tumor cells show immunoreactivity for muscle-specific proteins such as myogenin, myoD1, desmin, and muscle-specific actin [5,9]. Electron microscopy and molecular study has also been reported to aid the diagnosis [5,7,10].
The majority of cytomorphologic findings observed in our case were consistent with histopathology and previous literature, except for the horseshoe-shaped nuclei. These characteristics may cause concern about the possibility of malignant lymphoma, but they often appeared multilobated and peripherally located, sharing morphological similarity with multinucleated cells observed in the histology specimen and wreath-like nuclei reported in ARMS [8]. Karyorrhexis seemed to reflect the effect of prior therapy.
An ancillary study could not be performed in our case due to the limited amount and scarce cellularity of the CSF specimen, and attempt to destain and restain the cytology slides for muscle markers was unsuccessful. Nevertheless, the diagnosis of metastatic RMS could be made based on the patient’s history, neuroimaging and CSF cytology which was well-correlated with the histopathology. In many cases, a CSF sample is of insufficient volume because the sample is small and often shared among laboratories for multiple tests [11,12]. Immunocytochemistry, electron microscopy or molecular study may aid the diagnosis of RMS with a cytology specimen, but the small sample size makes these tests difficult in CSF. Therefore, it is important to be aware of the cytomorphologic characteristics, compare with histology, and to consider clinical history and neuroimaging findings in making the diagnosis.
The present case was initially diagnosed as ERMS, but the biopsy from the recurrent lesion was diagnosed as ARMS based on the IHC results. In the pathology review, myogenin showed diffuse strong immunoreactivity in both initial and recurrent lesions and features of ARMS, such as fibrovascular septa, were more evident in the neck dissection specimen (Fig. 2D, E). These findings support the diagnosis of ARMS over ERMS. A differential diagnosis of ARMS and ERMS can be challenging in small specimens or between solid variant of ARMS and ERMS with dense pattern, but myogenin immunostaining can provide diagnostic clues by showing diffuse, strong expression in ARMS and a more heterogeneous staining pattern in ERMS [13,14].
The RMS subtype is closely related to the prognosis and there have been several attempts to subtype RMS cytologically, primarily in the FNA specimens. After studying 37 FNA and six touch imprint samples, Atahan et al. [15] reported that cells with abundant cytoplasm and eccentric nuclei, multinucleated cells and background mucoid substance support the diagnosis of ARMS while tadpole or ribbon-shaped cells and small round cells support the diagnosis of ERMS. Findings more suggestive of ARMS were observed in our case and several other reports regarding ARMS in body fluid [5,8,10]. However, elongated cells of ERMS are rarely appreciated in body fluid, and we were unable to find literature that documented mucoid substance in fluid cytology of RMS [9]. These findings suggest that cytologic features that are characteristic of specific RMS subtypes can be less evident, and subtyping of RMS might be more challenging in body fluid. Thiryayi et al. [5] reported a case of metastatic ARMS in pleural effusion that showed diffuse, strong nuclear positivity for myogenin, which was confirmed by PAX3-FKHR gene fusion in FISH, however this report was not within the context of subtyping. It seems likely that myogenin immunostaining may aid subtyping in cytology specimens as in histopathology, but the utility needs to be validated in further studies.
ALK aberration is reported in both ARMS and ERMS, and is known to be associated with metastatic disease and poor survival in ERMS [16]. In our case, the neoplastic cells showed ALK positivity in IHC which correlated with a gain of ALK gene copy number in FISH. The patient received crizotinib, the ALK inhibitor, but the disease markedly progressed despite therapy. Crizotinib is approved for the treatment of ALK-rearranged non-small cell lung cancer, but the majority of ALK gene aberration in RMS is copy number gain [16]. There are limited clinical trial data on the effect of crizotinib on ALK-positive RMS and its efficacy on RMS requires further investigation [17].
Here we report a case of ALK-positive ARMS which invaded the CNS and developed multiple LMs. To our knowledge this is the first report to describe the cytologic features of RMS in CSF. RMS belongs to the small round blue cell tumors, and in the diagnostic process, the differential diagnosis of other small round blue cell tumors should be considered. Ancillary tests may aid in the diagnosis of RMS but are often difficult to perform in a CSF specimen. Therefore, it is crucial for cytopathologists to be aware of the cytologic features. It is also important to compare the cytology with histology, if available, and clinical history and neuroimaging findings should be considered in the diagnosis.
 XML Download
XML Download