

 PDF
PDF Citation
Citation Print
Print


Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytosis (LCH) of unknown etiology. It was first described in 1930 by the pathologist William Chester in the laboratory of Jakob Erdheim in Vienna, and over 500 cases have been reported in the published literature [1,2]. ECD is usually diagnosed between 40 and 70 years of age, and it has a slight male predominance [3]. In Korea, the first ECD case was reported in 1999, and fewer than 20 cases have since been reported [2]. ECD is a multisystem disease involving the long bones, cardiovascular organs, lungs, central nervous system, retroperitoneum, and skin [3-5]. The clinical presentation of this disease can vary strikingly but most commonly involves the skeletal system with bone pain (90%). There is usually symmetrical cortical osteosclerosis of the long bones and neurological involvement (50%) manifested as diabetes insipidus, cerebellar ataxia, and panhypopituitarism [1]. Other manifestations include cardiovascular involvement with pericardial pain or cardiac failure, as well as renal involvement and constitutional symptoms such as fever, fatigue, or weight loss [1]. Involvement of the lymph nodes or liver is rarely reported [6,7]. We report a rare case of ECD involving multiple systemic lymph nodes, pleura, liver, and the axial and appendicular skeleton.
CASE REPORT
A 53-year-old man was referred to the department of oncology for evaluation of rapid weight loss with inguinal lymphadenopathy. He experienced 20 kg of weight loss over 3 months, febrile sensation, night sweats, and general weakness. The patient had recently been diagnosed with diabetes mellitus, but had no other relevant medical history. Initial white blood cell (WBC) count was 4,790 cells/mm3 (neutrophil count 82%, lymphocyte count 5%), hemoglobin was 9.9 g/dL (normal range, 13.0 to 18.0 g/dL), and platelet count was 369,000/mm3 (normal range, 150,000 to 450,000/mm3). Erythrocyte sedimentation rate and C-reactive protein were 76 mm/hr (normal range, 0 to 15 mm/hr) and 7.59 mg/dL (normal range, 0.01 to 0.47 mg/dL), respectively. Other laboratory findings including urinalysis were nonspecific.
Abdominal computed tomography (CT) showed multiple enlarged lymph nodes involving the left para-aortic and aortocaval lymph nodes and common, external, and internal iliac lymph nodes (Fig. 1A–C). Splenomegaly and hepatomegaly were also suspected (Fig. 1B, D). Chest CT showed supraclavicular and mediastinal lymph node enlargement with an enhanced soft tissue mass involving the pleura (Fig. 1E, F). Positron emission tomography–computed tomography (PET-CT) was highly suggestive of malignant lymphoma involving systemic lymph nodes, consistent with CT images (Fig. 1G). PET-CT also indicated extensive involvement of the axial and appendicular skeleton including the skull, almost all vertebrae, both scapulae, pelvic bones, rib cage, sternum, and both femurs (Fig. 1H). The right pleura and spleen were also suspicious for lymphoma involvement. On bone scan, a mild and patchy activity increase was noted in the proximal shafts of both femurs and the left humerus. Subtle inhomogeneous activity was noted in the axial skeleton, although no significant activity was present. Based on imaging, malignant lymphoma with extensive systemic involvement was suspected. Additional laboratory studies showed β2-microglobulin of 4.115 μg/mL (normal range, 0.60 to 2.36 μg/mL), ferritin of 2,094 ng/mL (normal range, 30 to 400 ng/mL), and lactate dehydrogenase of 612 U/L (normal range, 250 to 450 U/L). Serum protein electrophoresis showed nonspecific findings.
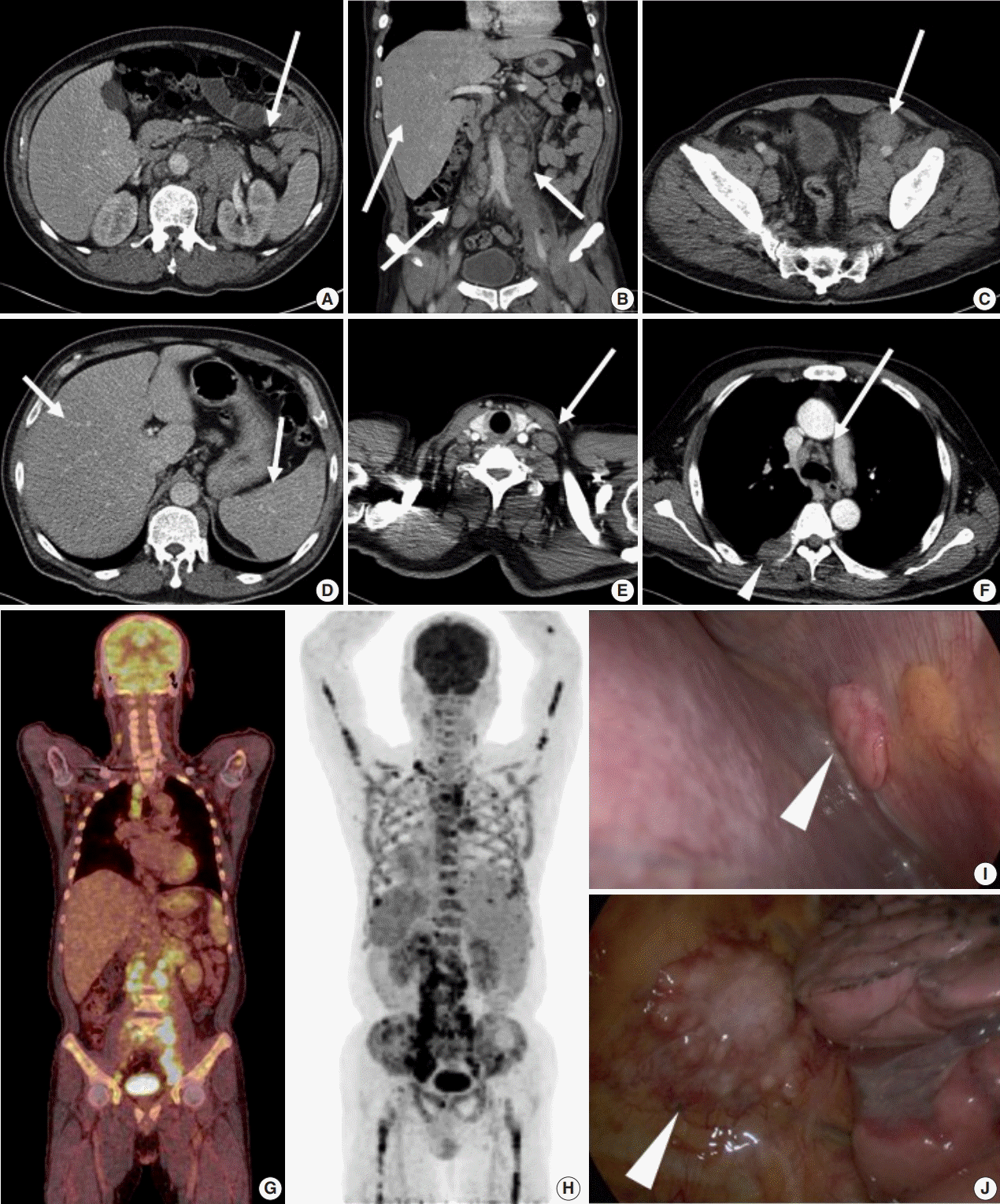 | Fig. 1.(A–C) Abdominal computed tomography (CT) scan shows multiple enlarged paraaortic, aortocaval, common iliac, and inguinal lymph nodes (arrows). (B, D) Hepatomegaly and splenomegaly are also suspected (arrows). (E, F) Chest CT scan shows supraclavicular lymph node enlargement (E, arrow) and mediastinal lymph node enlargement (F, arrow). (F) Pleural mass is detected on chest CT (arrowhead). (G) Positron emission tomography–CT scan shows enlargement of multiple mediastinal and retroperitoneal lymph nodes. (H) Diffuse axial and appendicular skeletal involvement was suspected. (I, J) Video-assisted thoracosurgery showed multiple pleural nodules attached to the parietal pleura (arrowhead).
|
For pathological confirmation, initial excisional biopsy of the right pleural mass was performed via video-assisted thoracosurgery. In the surgical field, multiple pleural nodules were attached to the parietal pleura with focal involvement of the pericardium (Fig. 1I, J).
Histopathological examination of the pleural biopsy revealed a proliferating lesion of spindle to oval-shaped cells with a fibrous background (Fig. 2A, B). Some cells showed vacuolated or focal clear cytoplasm. Otherwise, they had abundant eosinophilic, fine granular cytoplasm (Fig. 2C, D), suggestive of histiocytes. In some areas, tumor cells showed a marked spindle shape with randomly arranged fascicles (Fig. 2E, F). Some lymphocytes and a few eosinophils infiltrated the tumor cells and fibrous background. Nuclei were mostly round to oval with fine chromatin and a noticeable nucleolus, showing an overall bland appearance. A few cells with hyperchromatic and pleomorphic nuclei were noted, and mitosis was rare (< 1/10 high power field) (Fig. 2D). At the boundary of the mass, tumor cells focally infiltrated the surrounding adipose tissue.
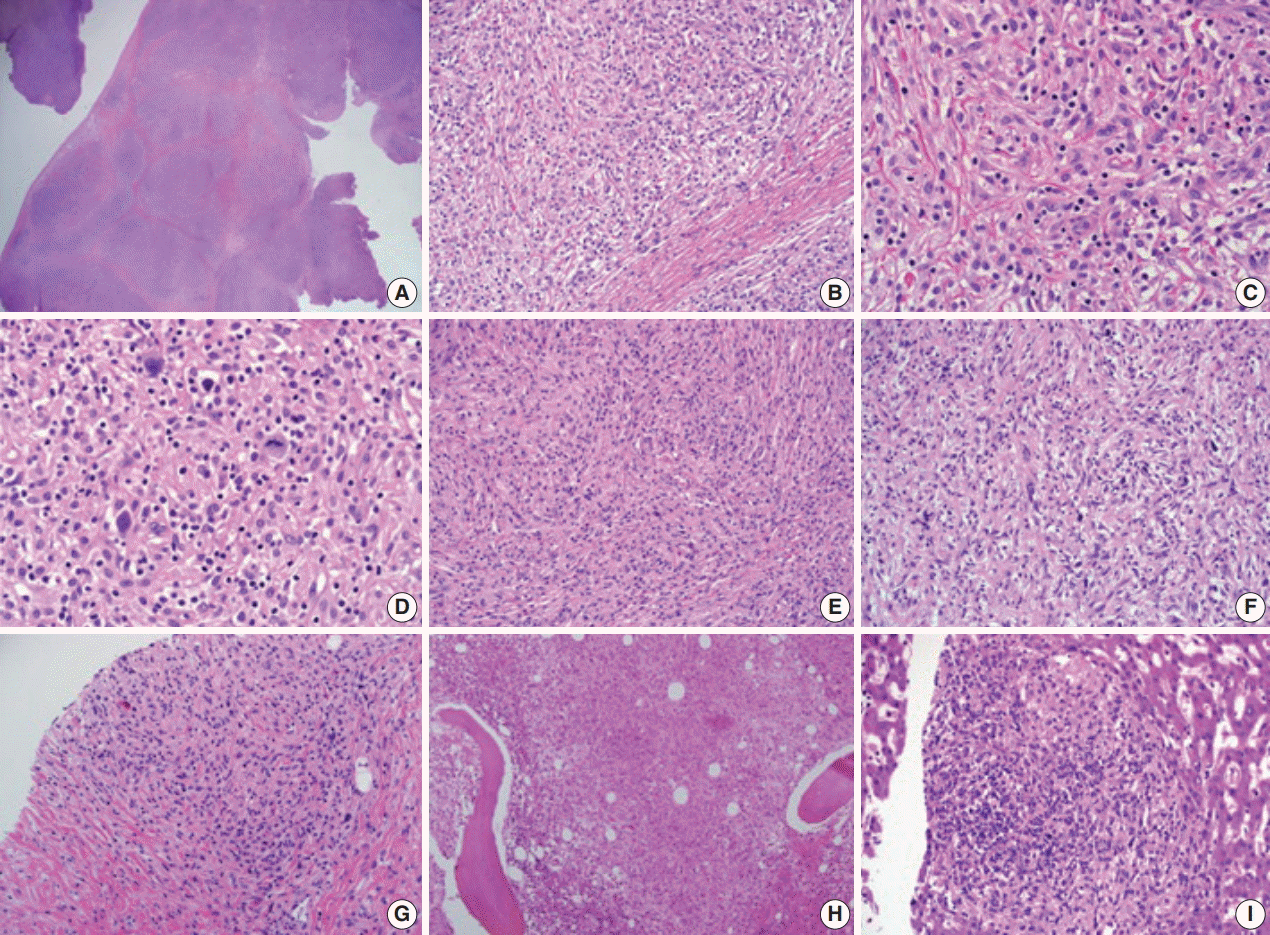 | Fig. 2.(A, B) Histopathologic examination of pleural biopsy revealed a proliferating lesion of spindle to oval-shaped cells with a fibrous background. (C) Some cells showed vacuolated or focal clear cytoplasm. Otherwise, they had abundant eosinophilic, fine, granular cytoplasm. (D) A few cells with hyperchromatic and pleomorphic nuclei were noted, and mitosis was rarely found (<1/10 high-power field). (E, F) In some areas, tumor cells showed marked spindle shape with the impression of randomly arranged fascicles. (G, H) External iliac lymph node (G) and bone marrow (H) biopsy showed similar tumor cells to the previous pleural biopsy. (I) Subsequent liver biopsy also revealed the same type of cells aggregating with fibrosis in the liver parenchyma.
|
With only these hematoxylin and eosin stain findings, the initial histopathological impression was spindle cell tumor, with a differential diagnosis of histiocytic neoplasm, dendritic cell sarcoma, sarcomatoid mesothelioma, and rare types of malignant lymphoma such as lymphocyte-depleted Hodgkin lymphoma. For further evaluation, immunohistochemical studies followed, including CD21, CD23, calretinin, epithelial membrane antigen (EMA), CD56a, CD45RB, CD30, CD15, Ki-67, and CD68 (Fig. 3A).
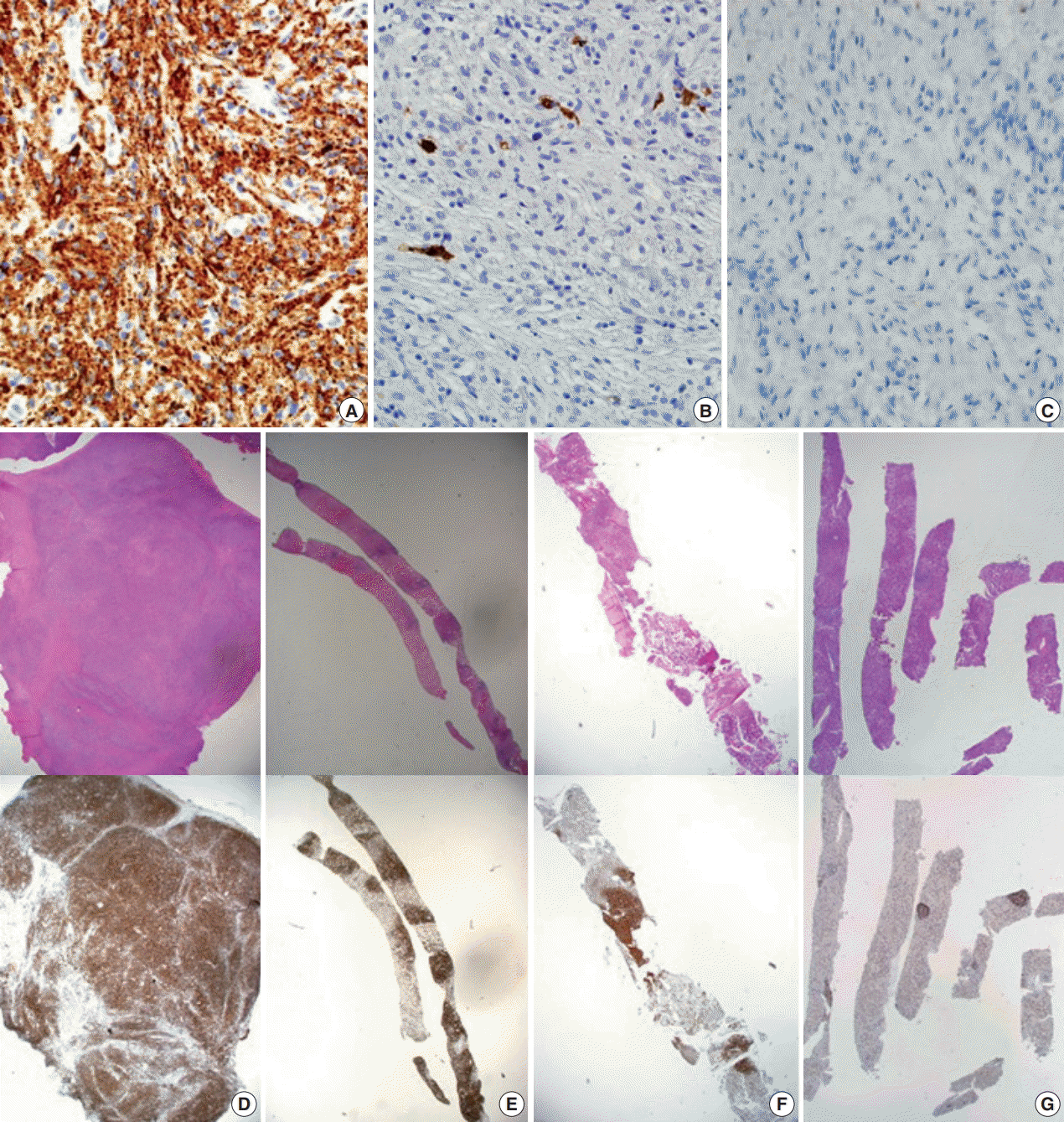
Dendritic cell sarcoma and sarcomatoid mesothelioma were excluded by negative findings for CD21, CD23, calretinin, and EMA along with low Ki-67 index (5%). Rare types of malignant lymphoma, such as lymphocyte-depleted Hodgkin lymphoma, were unlikely because of absence of reactivity for CD30, CD15, and CD45RB along with negative Epstein-Barr virus encoded RNA in situ hybridization (EBER-ISH). In addition, focal atypical lymphoid cells and some atypical Reed-Sternberg-like cells (RS-like cells) raised the possibility of peripheral T-cell lymphoma with RS-like cells; however, reactive T lymphocyte patterns of CD3, CD4, CD8, CD2, CD5, and CD7, as well as negative reactivity for EBER-ISH were found. T-cell receptor (TCR) gene rearrangement studies with polyclonal T-cell receptor δ (TCRD), γ (TCRG), and β (TCRB) results ruled out the possibility of malignant lymphoma. Instead, the tumor cells were uniformly positive for CD68, which is highly suggestive of a histiocytic lesion. Additional immunohistochemical studies of S-100 and CD1a were negative (Fig. 3B, C).
Rapid growth of the external iliac lymph node was detected on physical examination, and follow-up abdominal CT scan revealed the rapid size increase of multiple lymph nodes. Needle biopsy of an external iliac lymph node was performed. In addition, bone marrow biopsy was performed to exclude any other malignancy that might cause extensive skeletal fluorodeoxyglucose (FDG) uptake (Fig. 1G, H). Both biopsies (Fig. 2G, H) revealed tumor cells similar to those shown on previous pleural biopsy, expressing the same immunoprofile of CD68 (+), S-100 (–), and CD1a (–). Along with fibrosis, tumor cells replaced the normal lymph node structure and bone marrow space (Fig. 3E, F).
Radiological and histopathological results suggested a proliferating histiocytic lesion extensively involving bones, replacing bone marrow space, forming a pleural mass, and involving systemic lymph nodes. The differential diagnoses included LCH, Rosai-Dorfman disease (RDD), ECD, and benign xanthogranulomatous inflammation. The possibility of LCH or RDD was considered low for the following reasons: (1) all spindle cells from biopsied sites were negative for CD1a and S-100; (2) characteristic histologic findings of LCH, such as large oval cells with complex grooved and folded nuclei, were not observed; (3) even in the lymph node, a frequent site of RDD involvement, the characteristic morphology of RDD including expansion of sinuses by large histiocytes and emperipolesis, was not identified. In addition, proliferation of similar cells was not confined to a particular organ or site, suggesting a systemic condition rather than localized reaction or inflammation. In conclusion, the patient was diagnosed with ECD with systemic involvement.
Additional testing for the BRAFV600E mutation by real-time polymerase chain reaction was performed with the pleural biopsy tissue and was negative. Brain magnetic resonance imaging (MRI) was negative for disease involvement. MRI of the heart was not performed, but an echocardiogram showed negative findings for pericardial involvement, cardiac wall involvement, valve involvement, or major vessel involvement. Furthermore, serial chest CTs for follow up of the pleura, lymph nodes, and bone lesions revealed no ECD involvement of the aorta or heart.
Interferon-α (IFN-α) was administered 3 times a week (3 × 10 IU subcutaneous injection). After 1 month of treatment, the patient complained of whole-body myalgia, general weakness, and depressed mood. The authors considered these symptoms signs of an adverse response to INF-α, and the injection was delayed for 2 weeks. During this rest period, a response evaluation was performed. Chest and abdominal CT scan showed stable disease based on Response Evaluation Criteria in Solid Tumors ver. 1.0. After 1 additional month of IFN-α administration, the patient revisited the hospital complaining of anorexia, nausea, and sustained general weakness. WBC, hemoglobin, and platelet counts were 6,290 cell/mm3 (normal range, 4,000 to 10,000 cell/mm3), 7.5 g/dL (normal range, 13.0 to 18.0 g/dL), and 250,000/mm3 (normal range, 150,000 to 450,000/mm3), respectively, and aspartate transaminase, alanine transaminase, total bilirubin, and direct bilirubin were 11 U/L (normal range, 14 to 40 U/L), 14 U/L (normal range, 9 to 45 U/L), 5.49 mg/dL (normal range, 0.47 to 1.58 mg/dL), and 4.41 mg/dL (normal range, 0.13 to 0.47 mg/dL). Abdominal CT showed progression of multiple lymphadenopathy and hepatomegaly without biliary obstruction. With findings indicating progressive disease, high-dose steroids (dexamethasone 40 mg) were administered for 3 days. However, total bilirubin and direct bilirubin levels consistently increased up to 16.91 mg/dL (normal range, 0.47 to 1.58 mg/dL) and 14.36 mg/dL (normal range, 0.13 to 0.47 mg/dL), respectively. Subsequent liver biopsy and histological findings revealed multifocal aggregations of CD68 (+), S-100 (–), and CD1a (–) tumor cells with fibrosis in the liver parenchyma (Figs. 2I, 3G), identical to the previous biopsies of the pleura (Fig. 3D), lymph node (Fig. 3E), and bone marrow (Fig. 3F). Despite subsequent high-dose steroid treatment, the patient experienced rapid progression and hepatorenal syndrome and died due to hepatic failure.
This study was approved by the Institutional Review Boards of the Catholic Medical Center Office of the Human Research Protection Program (KC16ZISE0433), which included review of the patient’s informed consent.
Go to : 
DISCUSSION
In this case, a 53-year-old male patient presented with constitutional symptoms and multiple enlarged lymph nodes mimicking malignant lymphoma. He was diagnosed with ECD based on histological findings and immunohistochemical studies. ECD is a rare disorder of unknown etiology characterized by CD68 (+), S-100 (–), and CD1a (–) non-Langerhans histiocytosis most commonly involving long bones such as the femur, tibia, and fibula [1]. Clinical manifestations of ECD vary greatly depending on the site of histiocytic infiltration [1,5].
Despite various clinical manifestations of ECD, patients almost always show long bone involvement detected by bone scan or PET-CT scan [1]. In addition to skeletal involvement, neurological, retroperitoneal, pulmonary, cutaneous, and cardiovascular involvement of ECD have been found in more than 20% of patients over the course of the disease. Characteristic imaging findings such as “coated aorta” or “hairy kidney” appear with cardiovascular and renal involvement, respectively [5]. Such findings did not exist in this case, except for FDG uptake and signal increases in both femurs on PET-CT and bone scan. These findings were also identified in nearly all vertebrae (cervical, thoracic, lumbar, and sacrum), the skull, both scapulae, the sternum, and both humerus bones. In contrast with LCH, which usually involves the craniofacial bones [8], the axial skeleton is classically spared in ECD, with predominant involvement of the lower limbs, suggesting that this case is unusual. In addition, extensive involvement of the lymph nodes, liver, and spleen is also very rare in ECD. Few cases of ECD with lymph node involvement have been reported [2,6]. This atypical manifestation obscured the clinical diagnosis of ECD. Extensive lymph node enlargement, involvement of the liver and spleen, and constitutional symptoms gave the clinical impression of malignant lymphoma.
The histopathological findings of ECD are characterized by xanthogranulomatous tissue infiltration with lipid-laden histiocytes [1], which characteristically express an immunoprofile of CD68 (+), CD1a (–), and S-100 (–) [1-4,8]. However, specific or strict histopathological criteria for ECD are not established, such as the degree of “lipid-laden” features or characteristics including nuclear pleomorphism and arrangement pattern. In this case, cells of many foci were spindle-shaped with a fascicle pattern, and most nuclei were relatively large, oval, and plump with fine granular chromatin and nucleoli. A few cells were pleomorphic with hyperchromatic nuclei and mitosis, although they were very sparse. Most cells had fine, granular, abundant eosinophilic cytoplasm and clear foamy cytoplasm on high-power examination. With these findings, possible diagnoses included dendritic cell sarcoma, as well as histiocytic infiltrating lesions such as LCH, RDD, and ECD [9]. Nuclear grooving, prominent eosinophilic infiltration, and definite emperipolesis were hard to identify, although there were abundant lymphocytes and a few eosinophils. Malignant lymphoma was initially suspected, but eventually deemed an unlikely possibility. Immunohistochemistry was negative for CD45RB, CD30, and CD15, excluding malignant lymphoma. Likewise, negativity for CD21, CD23, S-100, CD1a, and calretinin along with low Ki-67 index excluded dendritic cell sarcoma, LCH (commonly CD1a [+]), RDD (commonly S-100 [+]), and other possible diagnoses such as undifferentiated pleomorphic sarcoma and sarcomatoid mesothelioma.
In this case, disease progression with hepatic involvement was suspected due to increases in total bilirubin and direct bilirubin level, which were proven by liver biopsy. The patient died four months after initial presentation. Previous literature has reported that ECD presenting with central nervous system (CNS) and cardiovascular involvement has a poor prognosis [10,11]. Data are lacking for ECD prognosis with lymph node or hepatic involvement, but our patient showed a poor prognosis over the course of treatment.
According to recently suggested revised classification, histiocytic disorders are grouped as L, C, R, M, and H by a system that incorporates histology, phenotype, molecular alterations, clinical findings, and imaging characteristics [12]. The “L” (Langerhans) group includes LCH, ECD, and indeterminate cell histiocytosis [12]. More than 80% of LCH and ECD cases express clonal mutations involving genes of the mitogen-activated protein kinase pathway [12]. In one study, about 20% of patients with ECD were found to also have LCH lesions [13]. Although no biopsy sites of our patient showed characteristic LCH morphology and were all negative for CD1a immunohistochemistry, the possibility of overlapping LCH histology cannot be ruled out, if other sites such as the vertebrae were also biopsied.
The largest cohort study to date identified BRAFV600E mutations in 46 of 80 ECD patients (57.5%) with multiple orthogonal methodologies [14]. In addition, some of the remaining BRAF wildtype patients had N/KRAS, PIK3CA, and AKT1 mutations [14]. Therefore, although our patient was negative for BRAFV600E mutation, the possibility of other genetic alterations found in previous studies of ECD cannot be ruled out. Unfortunately, further studies for other possible genetic alterations could not be performed due to the patient’s rapid disease progression and death.
There are few prospective therapeutic studies on ECD and no randomized controlled trials on ECD. Therefore, therapeutic guidelines have not been established. In general, IFN-α has been the drug of choice since the first report of its efficacy in 2005 [8]. Multivariate retrospective analysis suggested increased survival with IFN-α [15]. Although the optimal dose of IFN-α has not been established, 3 million units (mIU) 3 times per week were initially recommended [15]. Several studies recommended long-term (up to 3 years) treatment with high-dose IFN-α (9 mIU 3 times/wk) in cases of CNS or cardiac involvement [8,15]. In 2012, a somatic gain-of-function mutation (V600E) in the protooncogene BRAF was detected in 54% (13 of 24) of patients with ECD [15]. This mutation leads to activation of the tumorigenic RAS-ERK pathway and is thought to play a key role in the pathogenesis of the disorder [16]. Treatment targeting this mutation has been tried, and the results of four ECD patients treated with BRAF inhibitors vemurafenib or dabrafenib have been reported [8,16,17]. There are no larger studies addressing several remaining questions, such as the optimal drug dose, treatment duration, and possible long-term effects. However, long-term treatment with BRAF inhibitor can eventually lead to treatment resistance based on alteration of the RAS-MEK-ERK pathway. Recently, there was a report of a patient who developed resistance to dabrafenib and showed additional clinical response after adding the MEK inhibitor trametinib [17]. This report implies the promising role of target inhibitors in treatment of ECD. Other than RAS-ERK pathway alterations, several studies found that proinflammatory cytokines such as interleukin (IL)-1, IL-6, and tumor necrosis factor α (TNF-α) are increased in ECD lesions [18]. A recombinant IL-1R antagonist anakinra, an anti–TNF-α antibody infliximab, and a humanized monoclonal antibody against the IL-6R tocilizumab have been suggested as second-line treatments [8]. The treatment effects of proinflammatory cytokine inhibitors have been reported as case reports with positive efficacy in clinical response. However, there is a recent report of 12 patients treated with anakira after IFN-α failure who showed variable treatment response without partial response of measurable tumors [18]. Based on prior case series, further modulation cytokine inhibitors should be considered as second line treatment. Imatinib mesylate, a tyrosine kinase inhibitor, has been successfully used in case reports [19]. Imatinib selectively targets KIT, BCR-ABL, and platelet-derived growth factor. Haroche et al. [10] found abundant expression of platelet-derived growth factor receptor (PDGFR-β) in some histiocytic lesions of ECD and related disorders, and they successfully treated six patients with ECD positive for PDGFR-β with imatinib. They suggest that tyrosine kinase inhibitors more specific for PDGFR-β could be promising drugs for treating patients with severe forms of ECD resistant to IFN-α therapy. In addition, corticosteroids and cytotoxic drugs such as vinca alkaloids, anthracyclines, and cyclophosphamide can be administered conventionally, but the effects of those drugs are not significant [8,15]. Radiotherapy and debulking surgery for patients with ECD have been reported, but the effects are unclear [8]. In our case, INF-α was administered for first-line treatment but showed no treatment response. After IFN-α, high dose steroids were applied for symptomatic control, but hepatic dysfunction progressed. Due to rapid progression of hepatic failure, other agents were not attempted. BRAFV600E mutation was not detected in our patient, so vemurafenib was not applied as a systemic treatment.
Although the etiology of ECD had been veiled for decades, recent advances in genetic understanding of the disease have provided important clues. There is abundant evidence that, in patients with ECD and “L” LCH [12], BRAFV600E mutations exist in both histiocytes and bone marrow cells [20]. An animal experiment with mice also strengthened the hypothesis that ECD and LCH are forms of inflammatory myeloid neoplasia [20]. Histopathological findings of ECD appear benign, but when involving critical organs, numerous previous studies have shown poor prognoses and high mortality rates. Because ECD can involve any organ, clinicians should be aware of the disease and include it on differential diagnoses in suspicious clinical settings, even when the likelihood is low. The final diagnosis can be confirmed by identifying histiocytic lesions via histological and immunohistochemical studies in the context of a fitting clinical presentation and radiologic findings.
In this report, we present a unique case of ECD with an unusual presentation characterized by involvement of systemic lymph nodes, axial and appendicular skeleton, liver, and pleura. More data and investigation regarding the cells of origin are necessary to further understand this disease and establish effective treatment for patients, particularly those with critical organ involvement, rapid progression, and high mortality, as in this case.
Go to :
 XML Download
XML Download