

 PDF
PDF Citation
Citation Print
Print


Ependymoma is the third most common neuroepithelial tumor of the childhood [1]. It is a circumscribed glioma consisting of uniform small cells with ependymal differentiation, commonly originates from the walls of the cerebral ventricles or spinal canal, and is mostly manifest in children and young adults [2]. Ependymomas account for 9% and 8% of intracranial and intraspinal neoplasms of all primary brain and central nervous system (CNS) tumors in ages 0–14 and 0–19, respectively [3]. Like other primary glioneuronal tumors, extracranial metastasis of ependymoma is extremely rare [4-11]. Korshunov et al. [12] reported that the incidence of extracranial metastasis in their cohort was 2% (5/258 cases). In this paper, we present a rare case of anaplastic ependymoma with widespread extraneural metastasis. This study abides by the World Medical Association Declaration of Helsinki recommendations and was approved by the Institutional Review Board of Seoul National University Hospital (IRB No. 1507-040-690). The patient agreed and signed the agreement.
CASE REPORT
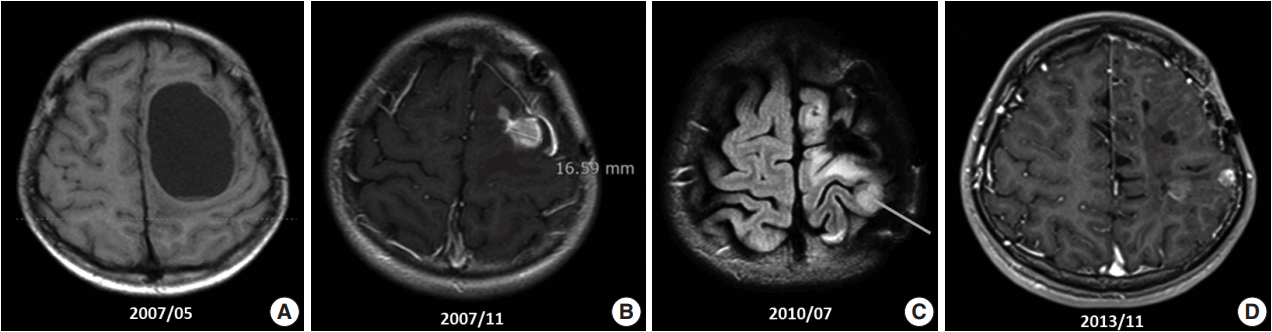
A 10-year-old boy visited the pediatric neurosurgery outpatient clinic due to headache and vomiting for 1 week. On physical examination, grade IV+ motor weakness in the right upper extremity and limping gait of the right leg were noted. Initial magnetic resonance imaging (MRI) revealed a large cystic mass (6.2 × 4.5 cm) in the left frontoparietal lobe (Fig. 1A). Craniotomy and gross total removal (GTR) of the tumor were performed in May 2007. Both solid and cystic portions were dissected from the normal-appearing brain parenchyma for GTR of the tumor. Histological diagnosis was World Health Organization (WHO) grade II ependymoma. Postoperatively, the patient’s neurological status was significantly improved, and right hemi-weakness gradually normalized.
Follow-up (F/U) MRI 3 months after initial surgery showed a small enhancing lesion with peripheral edema at the superolateral side of the operation bed compared with immediate postoperative MRI. At that time, clinicians could not rule out tumor recurrence. Therefore, the patient was treated with 8 weeks of adjuvant radiotherapy with 61.2 Gy. Five months after surgery, when the dose of antiepileptic drug was decreased, focal seizure developed as twitching in the right arm. After reverting to the full dosage of antiepileptic drugs, the symptoms disappeared. By that time, the right upper extremity weakness had almost normalized. However, 6 months after surgery, F/U MRI (Fig. 1B) revealed an increased extent of the recurrent tumor and peritumoral edema at the operation site compared with the 3-month F/U MRI finding (1 × 0.8 × 1.9 cm vs 1.5 × 1.6 × 3.1 cm). On reoperation 8 months after initial surgery, intraoperative ultrasound was used for GTR, and anterior, posterior, and both lateral margins were checked through frozen section biopsy. The second reoperation revealed WHO grade III anaplastic ependymoma. Subsequent F/U MRI was performed, and GTR was suggested.
Two months after the second operation, F/U MRI showed no definite enhancing recurrent mass; however, rim enhancement was noted along the resection margin. Tumor recurrence was suspected, and chemotherapy was considered; however, no recommendable chemotherapy regimen was available. Therefore, gamma knife surgery (GKS) (No. 1) was performed (volume, 3.7 cc; dose, 18 Gy at 52%; shot, 8 ×). However, the lesion enlarged again one month after the GKS. Subsequent GKS was then performed twice (GKS No. 2: volume, 1 cc; dose, 16 Gy at 50%; shot, 6 ×; GKS No. 3: volume, 1.8 cc; dose, 19 Gy; shot, 6 ×). Approximately a year after the second surgery, the tumor showed a rapid growth (February 2009, 13.8 × 18.6 × 13.8 cm; April 2009, 20.6 × 21.5 × 15.8 cm), and reoperation for GTR was considered but eventually could not be carried out. Without reoperation, there was a gradual decrease in tumor size for a year.
However, findings of the 2-year and 6-month F/U MRI worsened. Several newly appearing enhancements were found in the left parietal and temporal lobes, left cranial nerves (CN) VII and VIII, and right CN V. Leptomeningeal seeding was also noted (Fig. 1C). GKS No. 4 was performed again. A third operation was again considered due to increased enhancing mass, but was refused by the guardian. Fractionated GKS No. 5 was performed instead. Numbness of the right arm and myoclonic movement accompanied by limb weakness and sensory change in taste developed. F/U MRI showed increased enhancing mass and increased edema compared with the MRI findings a month ago. Fractionated GKS No. 6 was then performed. Right hemiparesis worsened. A third craniotomy and tumor removal was performed, and the tumor along the gliotic plane was totally removed. However, after surgery, right hemiparesis worsened; as such, the patient started to receive rehabilitation management. Two-month F/U MRI showed an increase in the size of the enhancing dura-based mass. Fractionated GKS No. 7 was done, which was preferred by the guardian. The patient then went to the emergency room due to right-sided tonic clonic seizure. On computed tomography scan, focal nodular enhancing lesion appeared again at the operation site. Tumor invasion to the incision scar and temporalis muscle was observed. Additionally, new enhancing nodule in the left parietal lobe was found (Fig. 1D). A fourth craniotomy and excision of the tumor, which showed enhancement on MRI, was subsequently performed.
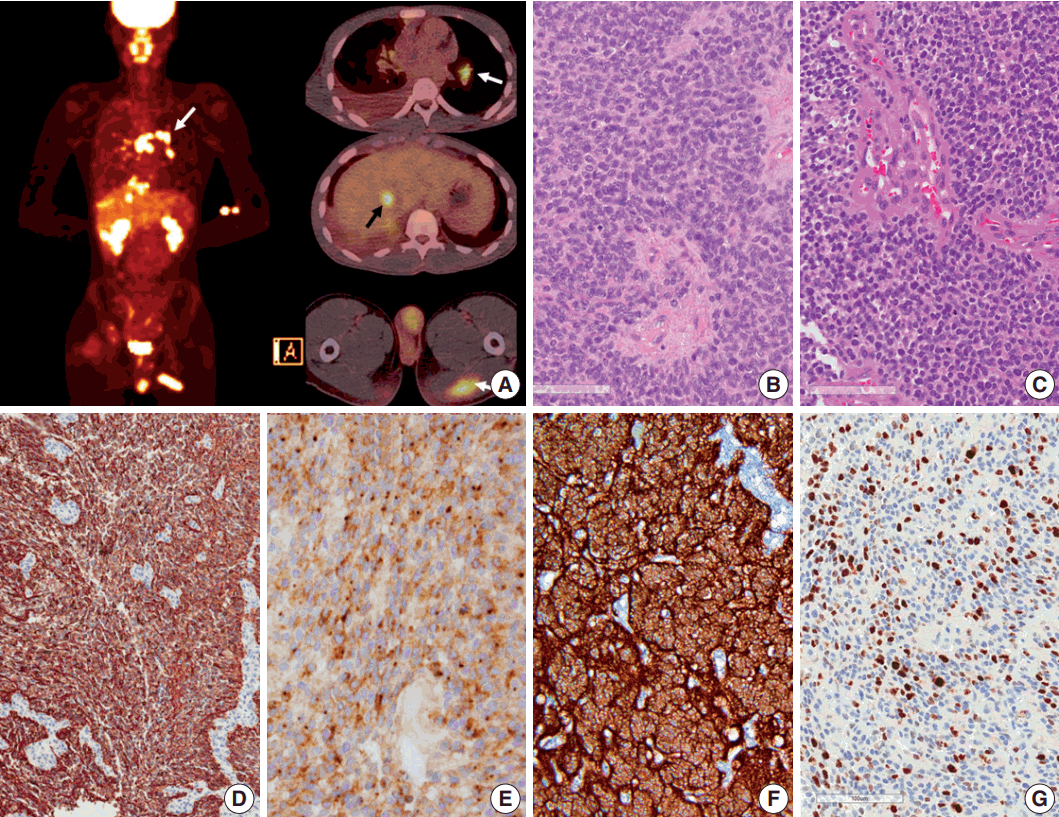
Seven years after initial presentation, several new tumors were found in the leptomeninges, scalp, temporalis muscle, lung, mediastinal lymph node (LN), liver, and bones on positron emission tomography imaging study (Fig. 2A). Excision of the scalp masses and needle biopsy of the liver were performed for diagnostic and palliative purpose (Fig. 3). The tumors were pathologically confirmed as mestastatic anaplastic ependymoma. The patient received chemotherapy, seven cycles of VIP (etoposide, ifosfamide and cisplatin).
However, the disease progressed and his condition worsened. He died after 9 years of having anaplastic ependymoma.
Pathological findings
Pathologic diagnosis of the initially resected tumor in 2007 was grade II ependymoma (Fig. 2B), with low mitotic rate (4/10 high-power field) and low Ki-67 labeling index (1%). However, the pathologic diagnosis of the second recurrence at 5 months after the initial GTR, which was removed 8 months after the initial GTR, was WHO grade III anaplastic ependymoma, with high mitotic rates, extensive necrosis, microvascular proliferation, and high Ki-67 (1:1,000, DAKO, Glostrup, Denmark) labeling index (Fig. 2C, G). Moreover, Ki-67 labeling indices in the specimen from leptomeningeal seeding were 24.5% to 37.3%, suggesting rapid malignant transformation. After GKS, the tumor showed extensive necrosis (more than 50%), suggesting that radiotherapy and GKS were effective to a certain level. However, the tumor eventually metastasized systemically. The tumor cells were robustly positive for glial fibrillary acidic protein (GFAP; 1:300, DAKO), and showed extensive dot-like positivity for epithelial membrane antigen (1:100, DAKO) (Fig. 2D, E). Results of the ultrastructural study showed microvilli and cilia in the intercellular and intracytoplasmic microrosettes. In addition, long intermediate junctions were present. Collectively, these ultrastructural findings suggested anaplastic ependymoma. In all specimens, L1CAM (1:200, Abcam, Cambridge, UK) was robustly positive in the entire tumor cell cytoplasm, suggesting RELA fusion-positive ependymoma (Fig. 2F).
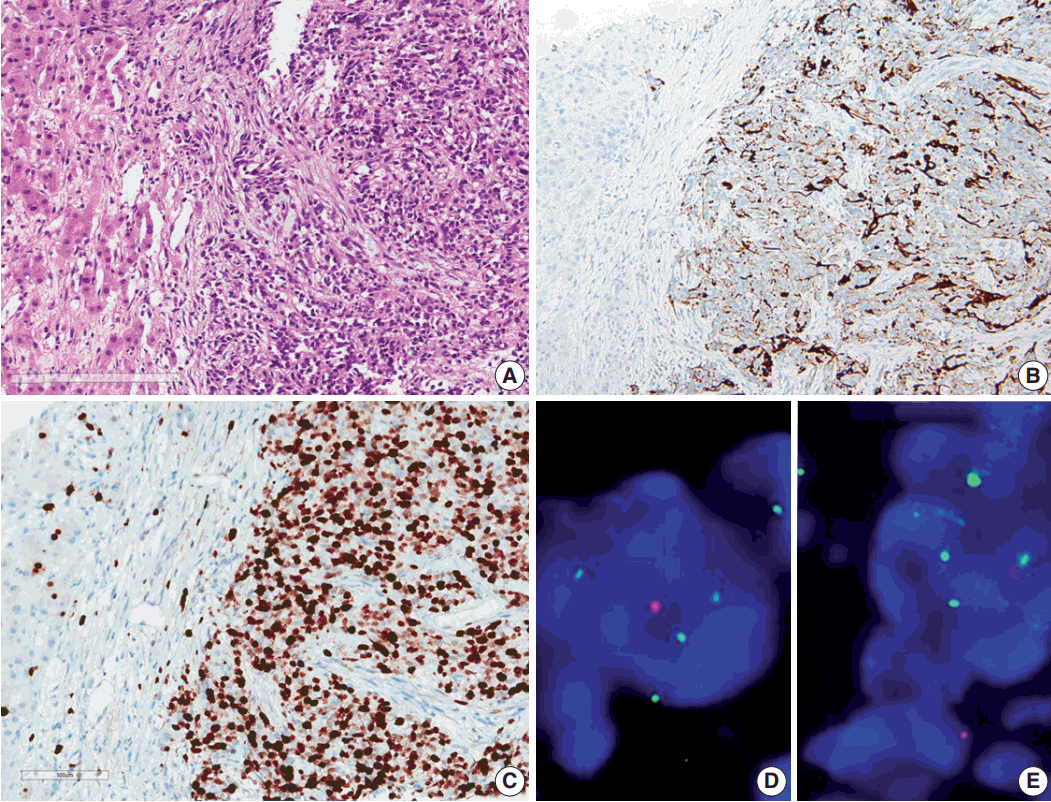
Results of the liver biopsy revealed metastatic anaplastic ependymoma, which showed robust GFAP positivity and high Ki-67 index of 92.47% (Fig. 3A–C).
Using formalin-fixed paraffin embedded tissue, fluorescence in situ hybridization (FISH) was performed to examine genetic characteristics. Locus-specific 1p36 (Spectrum Orange)/LSI 1q25 (Spectrum Green) dual-color FISH Probe (Vysis, Downers Grove, IL, USA) and 9p21.3 (CDKN2A) (Spectrum Orange)/CEP9 (Spectrum Green) dual color FISH Probe (Vysis) were used. A total of 100 nuclei were counted. The 1q25/1p36 ratio was 1.55, which revealed 1 copy gain of chromosome 1q25, and 9p21.3 homozygous deletion was also found (Fig. 3D, E).
DISCUSSION
Ependymomas are tumors arising from ependymal lining cells, which are classified according to the location and age of the patient because different genetic abnormalities and biological behaviors are found according to these two parameters [13,14]. Ependymomas are more common in children than in adults and are the third most frequent neuroepithelial tumors of childhood [1]. Ependymomas in children tend to occur in the posterior fossa (PF), whereas they tend to occur in the supratentorium and spinal cord (SC) in adults. The tumors can be graded as WHO grade II and III according to histopathological parameters, such as mitotic rate, microvascular proliferation, and nuclear pleomorphism; however, it is difficult to apply and the clinical utility is questionable [15].
By far, no effective anticancer regimen has been found against ependymomas. Surgical resection and radiotherapy is the treatment of choice. Therefore, unlike other CNS gliomas, the outcome of ependymomas does not follow grade but the location and resectability.
Ependymomas are divided into three groups according to their location: supratentorial (ST), PF, and SC. ST ependymoma is further categorized as RELA-C11 or f95 fusion and YAP1-MAMDL1 or YAP1-FAM118B fusion [16]. RELA fusion subtype comprises 70% of ST ependymomas, and the rest is YAP1 fusion [16]. The RELA fusion group has remarkably worse survival than that of YAP1 fusion group. RELA fusion can be detected via reverse transcriptase–polymerase chain reaction or immunohistochemical marker L1CAM [15]. In case of PF ependymomas, LAMA2-expressing ependymomas (group A) has worse outcome than NELL2-expressing ependymomas (group B) [17].
At initial presentation, the tumor is located in the left frontoparietal area. In general, patients with ST ependymomas are believed to have better survival rates than those with infratentorial ependymomas because GTR is possible [18]. However, several contradictory reports regarding patients’ outcomes have been published [19].
The most important prognostic factor for ependymomas is genomic aberration. In cases of ST ependymomas, RELA fusion-positive ependymomas have worse prognosis than YAP1 fusion-positive ependymomas [8]. Additionally, gain of 1q25 and homozygous deletion of CDKN2A (9p21.3) are powerful independent indicators of unfavorable prognosis [13]. Our case is L1CAM-expressing ST ependymoma with poor clinical course and extracranial metastases to the skull, temporalis muscle, lung, mediastinal LN, liver, and bone.
Rickert published a review article on extracranial metastasis of pediatric brain tumors that included six cases of ependymoma, not otherwise specified, and two cases of anaplastic ependymoma [20]. Among them, one had metastasis without prior surgical intervention, including biopsy, and seven had metastasis after surgical intervention. The mean latency, which is the latency between brain surgery and extracranial metastasis, for metastasis was 25.7 months for non-shunt related metastasis.
In our case, although three surgeries had been done before extracranial metastasis, the latency for extracranial metastasis was 78 months from the first surgery. We assume that the surgery might have played a major role in metastasis to the skull and temporalis muscle; however, the tumor recurrence in the primary site despite GTR in a short duration (in 8 months) and repeated recurrences suggest its aggressive nature, which might be due to its genetic alteration of RELA fusion positivity and 1q25 gain. In general, cerebrospinal dissemination indicates poor prognosis in brain tumors [15]. Our case showed leptomeningeal seeding after tumor recurrence in the primary site.
In this paper, we report a case of aggressive anaplastic ependymoma with extracranial metastasis, positive for RELA fusion, 1q25 gain, and CDKN2A homozygous deletion.
 XML Download
XML Download