

 PDF
PDF Citation
Citation Print
Print


Helicobacter pylori is a common gastric pathogen that causes gastritis, peptic ulcer disease, gastric adenocarcinoma, and mucosa-associated lymphoid tissue (MALT) lymphoma. H. pylori infection has been linked to a number of rare gastric mucosal lesions with distinctive histological features, including rare forms of gastritis, such as lymphocytic gastritis (LG) [1-5] or granulomatous gastritis (GG) [3,4,6,7], and abnormal immunoglobulin deposits, such as Russell body gastritis (RBG) [8,9] or crystal-storing histiocytosis (CSH) [10,11]. These lesions are easily diagnosed based on their distinctive histological features; however, their development can be attributed to various factors besides H. pylori infection. Therefore, the role of H. pylori as a causative organism remains debatable, and it has been suggested that H. pylori may be an “innocent bystander [2,4,12].” Nevertheless, H. pylori eradication therapy has been linked to clinical and histological improvements in a subset of these lesions [7,13-21], which supports the role of H. pylori infection in their development and implies that H. pylori eradication therapy could be an effective treatment. Therefore, in this review, we investigate the relationship between these rare gastric lesions and H. pylori infection, describe their characteristic histological features, and evaluate the role of H. pylori infection in their pathogenesis.
RARE FORMS OF GASTRITIS
LG and GG are not distinct clinicopathological entities, but rather morphologic patterns of injury that can be secondary to a variety of underlying etiologies [2-4,6]. The histological identification of intraepithelial lymphocytosis and granuloma formation are key diagnostic features of LG and GG, respectively [1-7]. However, a morphological diagnosis of LG and GG should elicit clinical and laboratory workups to identify the underlying etiology. In Western countries, LG and GG are generally classified as special forms of H. pylori–negative gastritis [3,4]. However, there is convincing evidence that H. pylori infection contributes to the pathogenesis of both LG [1,5] and GG [6,7,22], and that H. pylori eradication therapy may be an effective treatment [7,13-15,17,23,24]. Thus, it is possible that subsets of LG and GG could be categorized as H. pylori–associated gastritis. The potential role of H. pylori infection in the pathogenesis of each of these lesions is described below, along with their histopathological characteristics.
Lymphocytic gastritis
LG, first described by Haot et al. [25] in 1988, is a rare form of chronic gastritis that is characterized by a dense lymphocytic infiltration of the surface and pit gastric epithelium known as “intraepithelial lymphocytosis.” LG was initially considered to be related to varioliform gastritis, which manifests as thickened mucosa with “octopus-sucker” targetoid erosions [25,26]; however, the endoscopic features of LG vary according to its severity, ranging from normal to hypertrophic [2,27]. Histologically, LG is defined by the presence of ≥25 intraepithelial lymphocytes (IELs) per 100 epithelial cells (Fig. 1). Gastric intraepithelial lymphocytosis is associated with a variety of conditions, including celiac disease, H. pylori infection, Crohn disease, syphilis, hypertrophic gastropathy, Ménétrier’s disease, human immunodeficiency virus, and lymphoma [2,5]. However, celiac disease and H. pylori infection are the main causes of LG, accounting for 38% and 29% of cases, respectively [5,13,28]. The association between LG and celiac disease is relatively well established. Mild intraepithelial lymphocytosis with a low cut-off value (≥8 IELs/100 epithelial cells) has been observed in 84% of patients with celiac disease [29], and LG occurs in up to 45% of patients, with resolution of LG in response to a gluten-free diet [5,13,28-30].
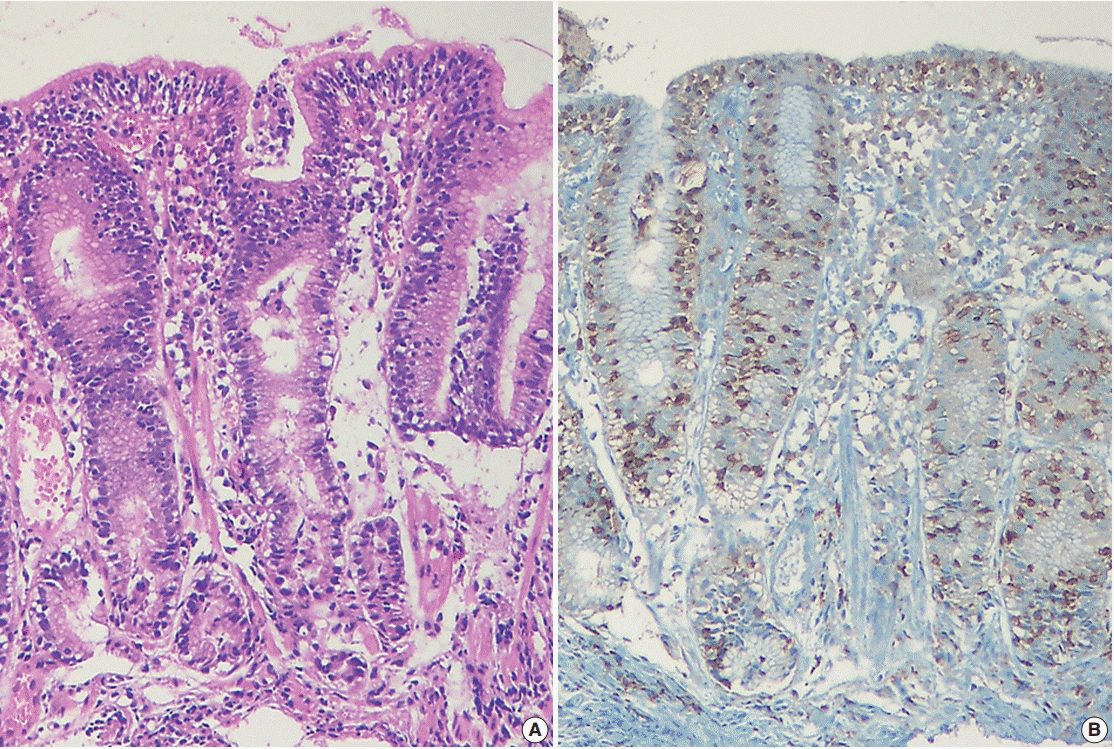
However, questions remain regarding the role of H. pylori infection in the development of LG due to the discrepancy between the prevalence of H. pylori in the general population and the incidence of LG among patients with H. pylori–associated gastritis [2-4]. Given the high global prevalence of H. pylori infection, it is unclear why the proportion of H. pylori–infected patients presenting with LG morphology (<5%) is so low [5]. Nevertheless, H. pylori eradication therapy can resolve H. pylori–associated LG by reducing IEL levels, and has been shown to improve symptoms and/or lead to regression of the gastritis [13-16], which supports a causal role of H. pylori infection in the development of LG. Previous studies have reported that H. pylori–associated LG frequently exhibits significant neutrophilic activity in addition to intraepithelial lymphocytosis [2,3,16,27], which is distinct from celiac disease-associated LG that exhibits intraepithelial lymphocytosis without neutrophilic infiltration. In this context, Nielsen et al. [27] argued that H. pylori–associated intraepithelial lymphocytosis accompanied with significant neutrophilic infiltration should be considered “chronic active gastritis,” rather than LG. However, considering the fact that LG is a morphological diagnosis that is based on the presence of intraepithelial lymphocytosis (≥25 IELs per 100 epithelial cells), the use of the term “LG” remains relevant.
Interestingly, a considerable number of patients with LG and positive H. pylori serology did not exhibit histological evidence of H. pylori infection [1,14,15,27]. In addition, the beneficial effects of H. pylori eradication therapy in LG have been observed in patients who had positive serology but negative histology for H. pylori [14,15]. Furthermore, even when H. pylori infection has been histologically confirmed in patients with LG, colonization tends to be mild and focal [14,15]. These results imply that failure to histologically detect H. pylori may be related to sampling error due to low-level infection [27]. Also, it is possible that LG development is a local, transient, or delayed immunological reaction to H. pylori infection, and is not a direct effect of the infection [1,13,14,27]. Thus, if other causes can be excluded, H. pylori eradication therapy could be considered in symptomatic patients with LG who are histologically and/or serologically positive for H. pylori. This approach may be more appropriate in countries with a low prevalence of celiac disease and a high prevalence of H. pylori infection, even without histological detection of H. pylori.
IELs are functionally and phenotypically distinct from peripheral lymphocytes, and the majority of IELs in the gastrointestinal epithelium are CD3+/CD8+ T cells with cytotoxic potential [31]. These IELs are thought to play an important role in mucosal immunity and have also been implicated in epithelial cell turnover by eliciting apoptosis through cytotoxic T-lymphocytes (CTLs) [31,32]. Recently, Han et al. [33] studied IEL subpopulations and their cytotoxicity in H. pylori–infected gastric mucosa using T-cell–restricted intracellular antigen-1 (TIA-1; a marker for resting and activated CTLs) and granzyme B (GrB; a marker for activated CTLs). They found that the IELs consisted of a mixture of TIA-1+/GrB– CTLs, TIA-1+/GrB+ CTLs, and CD4+ T cells in the infected mucosa. In addition, they found that H. pylori–associated LG was distinct from H. pylori gastritis, based on the increased IEL levels and changes in the cytotoxicity and distribution of the subpopulations: H. pylori–associated LG had a higher proportion of activated GrB+ CTLs, compared to H. pylori gastritis. There was also a parallel increase in epithelial apoptosis. Meanwhile, in a study by Oberhuber et al. [34], the proportion of GrB+ CTLs in H. pylori–associated LG (10.8%) was lower than that in idiopathic LG (12%) or celiac disease–associated LG (18.9%). Thus, although LG exhibits consistent histological features (regardless of etiology), IEL characteristics may vary depending on the underlying condition, which can lead to different clinical manifestations.
Granulomatous gastritis
GG is a rare disease that is characterized by the presence of granulomas, and is detected in 0.01%–0.35% of gastric biopsies [6,7,17,35]. GG can be caused by a number of factors, including systemic disease (e.g., Crohn disease, sarcoidosis, or vasculitis), infection (e.g., tuberculosis, histoplasmosis, or syphilis), underlying malignancy, or foreign bodies [4,6,7,36]. Crohn’s disease and gastric sarcoidosis are the two leading causes of GG, accounting for 20%–50% of cases in Western countries [6,35]. Isolated or idiopathic granulomatous gastritis (IGG) was first described by Fahimi et al. [37] in 1965, and is diagnosed by the exclusion of other granulomatous diseases. However, whether IGG can be considered a discrete condition remains controversial, as it is possible that a clear etiology could be identified through a more meticulous clinical work-up and long-term follow-up.
Dhillon and Sawyerr [22] first reported an association between GG and H. pylori infection in 1989. Since then a number of reports have been published that support their findings [6,7,17,18,23,24,38-42]. These reports demonstrated that the mucosa surrounding granulomas in GG exhibits typical histological features of H. pylori gastritis, that features suggestive of other etiologies are absent, and that H. pylori eradication therapy can result in GG resolution. However, whether H. pylori plays a causative role in the pathogenesis of GG remains debatable. First, the incidence of GG is abnormally low relative to the H. pylori prevalence in the general population [6,7,12,35]. Although Ectors et al. [6] and Maeng et al. [7] have reported the presence of H. pylori in 92% and 89% of GG cases, respectively, the overall incidences of GG were only 0.27% and 0.08%, respectively. Second, H. pylori organisms are rarely found within granulomas, implying H. pylori infection is a comorbidity rather than a cause in GG pathogenesis. Lastly, in cases of GG with H. pylori infection, granulomas often persist for 3–17 months after H. pylori eradication therapy [17,24,39], making its efficacy in GG questionable. It is plausible that although H. pylori can cause GG, granuloma formation is the result of a rare host response as opposed to a direct effect. Thus, H. pylori eradication therapy would be less effective in the resolution of GG, compared to its efficacy in conventional H. pylori gastritis.
Histologically, H. pylori–associated GG exhibits small non-necrotizing epithelioid granulomas with Langhans giant cells (Fig. 2), which are similar to those that are associated with sarcoidosis or Crohn disease. These granulomas tend to form in distinctive locations, such as the foveolar isthmi [6], with Maeng et al. [7] reporting that the majority (66.7%) of granulomas were found there. They are also often in contact with a damaged pit (where H. pylori are commonly found) and are frequently accompanied by prominent neutrophilic infiltration, which distinguishes them from the granulomas that are observed in Crohn disease or sarcoidosis [6,7]. However, this characteristic morphology and localization has not been consistently reported in subsequent studies [18,23,24,40-42]. Therefore, there are no distinct histologic features that could be used to confirm H. pylori–associated GG. Rather, it appears that the conditions in the mucosa surrounding granulomas are more informative [3,4,7], as the presence of H. pylori and neutrophil-rich chronic active gastritis (associated with glandular atrophy or intestinal metaplasia) increase the likelihood of H. pylori infection. Taken together, although the presence of H. pylori in the vicinity of granulomas does not imply a causative association, H. pylori eradication therapy should be considered when there are no other underlying disease except H. pylori infection.
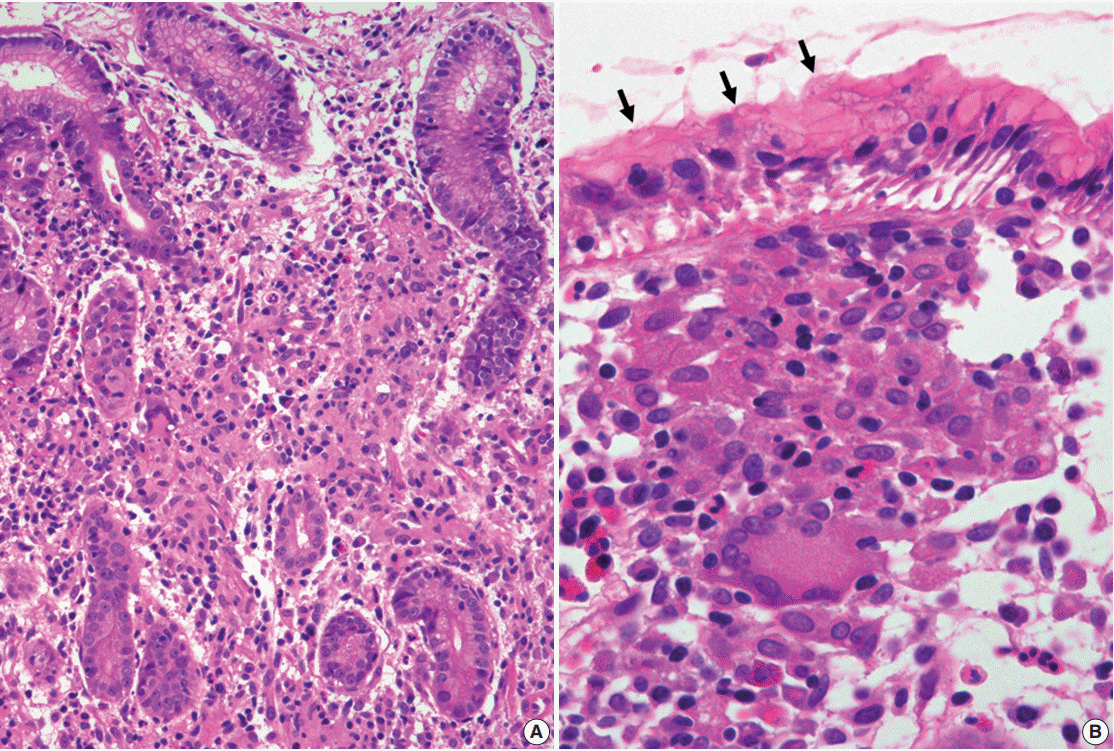 | Fig. 2.Granulomatous gastritis. (A) The biopsy specimen demonstrates diffuse chronic active gastritis with confluent granulomas including multinucleated giant cells. (B) A well-defined granuloma is noted just below the surface foveolar epithelium. Some Helicobacter pylori organisms are seen (arrows).
|
Go to : 
INTRACELLULAR IMMUNOGLOBULIN ACCUMULATION IN ASSOCIATION WITH HELICOBACTER PYLORI INFECTION
Immunoglobulin accumulations can be found in reactive and neoplastic plasma cells and include large intracytoplasmic spherical inclusions (Russell bodies), small intracytoplasmic morular inclusions (Mott cells), intranuclear inclusions (Dutcher bodies), and rare angular- or needle-shaped intracytoplasmic crystalline inclusions. These accumulations are associated with chronic inflammation with plasmacytosis, autoimmune disease, multiple myeloma, or other B-cell lymphomas [43-45]. Although the exact mechanism of immunoglobulin accumulation is unclear, it may be due to simple over-production, altered production, abnormal secretion, or impaired excretion [46,47]. In the gastric mucosa, diffuse plasma cell infiltration with immunoglobulin overproduction may result from chronic over-stimulation of plasma cells by mucosal pathogens, especially H. pylori. Scattered Russell bodies are often observed with H. pylori gastritis, whereas Dutcher bodies are frequently associated with low-grade MALT lymphoma. However, RBG and CSH are rarely reported in the stomach [9,10]. As the incidence of these lesions is low relative to H. pylori–associated gastritis, the contribution of H. pylori infection to their development is questionable. Furthermore, careful evaluation of their underlying cause is essential, as they can be associated with monoclonal gammopathy or lymphoreticular neoplasms [20,43,48]. The possible connections between H. pylori infection and these immunoglobulin accumulations, diagnostically relevant histological features, and biological significance are described below.
Russell body gastritis
The first case of RBG was reported by Tazawa and Tsutsumi in 1998 [19]. RBG is a rare form of chronic gastritis characterized by localized accumulation of Mott cells, which are plasma cells with a cytoplasm packed with small spherical inclusions (Fig. 3A, B). These lesions are rare, and only 30 cases of RBG have been published to date in the English literature [8,9,19-21,48-61]. The clinical and pathological features of RBG are summarized in Table 1. Although its pathogenesis has not been fully elucidated, there is evidence to support a strong association between H. pylori infection and RBG development. For example, H. pylori is detected in approximately two-thirds of patients with RBG [8,9,19-21,48,50,52,53,55,57,60,61]. Few other infections that have been reported with RBG include human immunodeficiency virus (three patients) [51,53,59], hepatitis C virus (one patient) [58], and candida esophagitis (one case) [49]. In addition, more than 60% of patients with RBG exhibit lesion regression following H. pylori eradication therapy [19-21,53,60]. Furthermore, it has been reported that highly virulent H. pylori genotypes (vacA and cagA) are associated with the formation of Russell bodies and Mott cells in the antral mucosa [62].
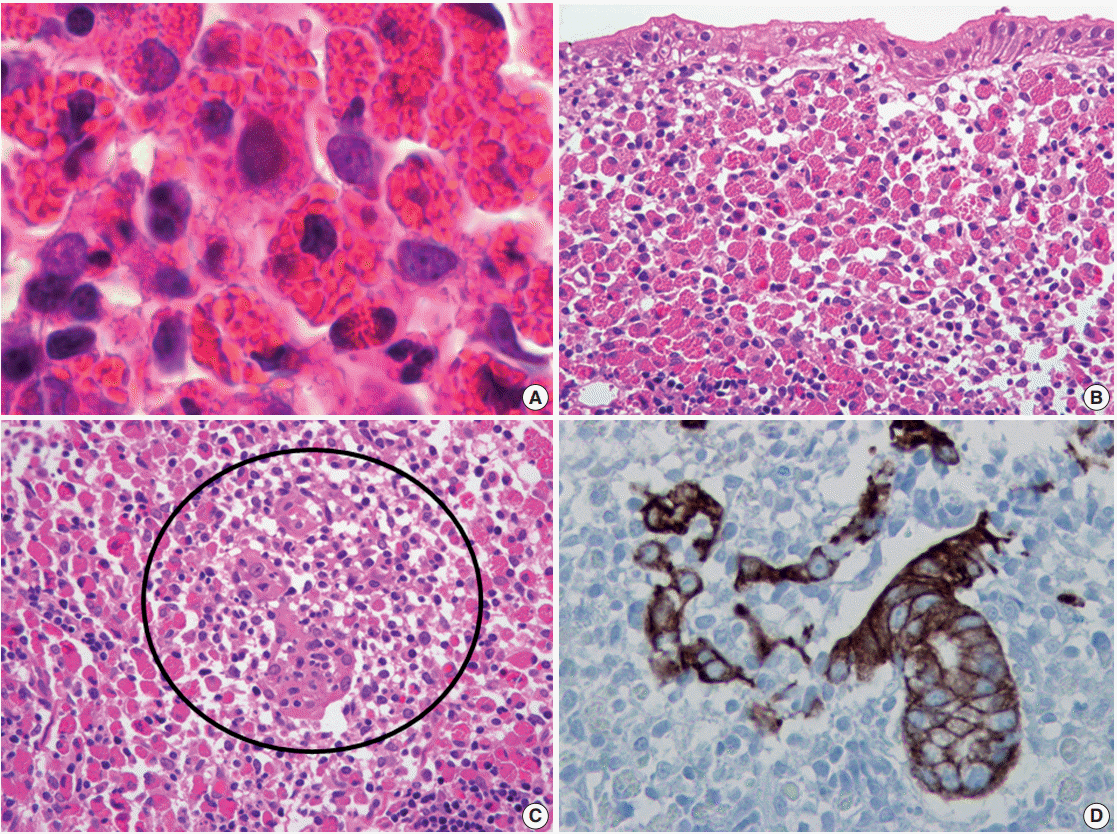 | Fig. 3.Russell body gastritis with concomitant mucosa-associated lymphoid tissue lymphoma. (A) Mott cells are plasma cells in which the cytoplasm is packed with multiple variable-sized Russell bodies. (B) The lamina propria of the gastric mucosa is expanded by extensive infiltration of Mott cells, consistent with Russell body gastritis. (C) Small- to intermediate-sized atypical lymphoid cells, morphologically consistent with centrocyte-like cells are admixed with Mott cells and destroy adjacent gastric glands to form a lymphoepithelial lesion (circle). (D) Immunostaining for cytokeratin highlights a lymphoepithelial lesion.
|
Table 1.
Clinical and pathologic findings of previously published cases of Russell body gastritis in the English literature
| Case | Study | Age (yr)/Sex | Endoscopic finding | Helicobacter pylori infection | Ig light chain of Mott cells | Gastric lesion coexisted | HPET/Resolution of RBG | Others |
|---|---|---|---|---|---|---|---|---|
| 1 | Tazawa and Tsutsumi [19] | 53/M | Multiple ulcer scars | Yes | Polyclonal | None | Done/Yes | - |
| 2 | Erbersdobler et al. [49] | 80/F | Irregular mucosal swelling | No | Polyclonal | None | NS | Candida esophagitis |
| 3 | Ensari et al. [50] | 70/M | Flattened gastric folds | Yes | Polyclonal | None | Done/NS | - |
| 4 | Paik et al. [8] | 47/F | Erythematous swelling | Yes | Polyclonal | None | Done/NS | - |
| 5 | Paik et al. [8] | 53/F | Yellowish raised lesion | Yes | Polyclonal | None | Done/NS | - |
| 6 | Wolkersdörfer et al. [20] | 54/M | Erythema and erosions | Yes | Monoclonal (λ) | None | Done/Yes | MGUS |
| 7 | Drut and Olenchuk [51] | 34/M | Elevation with central macule | No | Polyclonal | None | NS | HIV infection |
| 8 | Pizzolitto et al. [52] | 60/F | Minute-raised granular areas | Yes | Polyclonal | None | Done/NS | - |
| 9 | Licci et al. [53] | 59/M | Hyperemia | Yes | Polyclonal | None | Done/Yes | HIV infection |
| 10 | Habib et al. [54] | 75/M | Nodular chronic active gastritis | No | Polyclonal | None | NS | - |
| 11 | Shinozaki et al. [55] | 74/M | Centrally ulcerated bulky mass | Yes | Polyclonal | EBV-positive carcinoma | No | - |
| 12 | Shinozaki et al. [55] | 29/F | Ulcerated mass | Yes | Polyclonal | EBV-positive carcinoma | No | - |
| 13 | Del Gobbo et al. [56] | 78/F | Hyperemic gastric mucosa | No | Polyclonal | None | No | - |
| 14 | Wolf et al. [57] | 67/M | Exophytic tumor | Yes | NS | Signet ring cell carcinoma | No | - |
| 15 | Coryne and Azadeh [58] | 49/M | Severe raised erosive gastritis | No | Monoclonal (κ) | None | NS | HCV infection |
| 16 | Bhalla et al. [59] | 82/M | Gastritis | No | Polyclonal | None | NS | HIV infection |
| 17 | Karabagli and Gokturk [60] | 60/M | Large ulcerofungating mass | Yes | Polyclonal | None | Done/Yes | - |
| 18 | Yoon et al. [21] | 57/M | Elevation with central depression | Yes | Polyclonal | None | Done/Yes | - |
| 19 | Yoon et al. [21] | 43/M | Whitish flat lesion with nodularity | Yes | Polyclonal | None | Done/Yes | - |
| 20 | Araki et al. [61] | 74/F | Ulcer | Yes | Monoclonal (κ) | None | No | - |
| 21 | Zhang et al. [9] | 78/F | Gastritis with uneven mucosa | No | Monoclonal (κ) | None | No | - |
| 22 | Zhang et al. [9] | 77/F | Gastritis with uneven mucosa | Yes | Monoclonal (κ) | None | No | - |
| 23 | Zhang et al. [9] | 77/F | Punctiform erosion | Yes | Monoclonal (κ) | None | No | - |
| 24 | Zhang et al. [9] | 56/M | Raised erosion | Yes | Monoclonal (κ) | None | No | - |
| 25 | Zhang et al. [9] | 76/M | Erythema | Yes | Monoclonal (κ) | None | No | - |
| 26 | Zhang et al. [9] | 50/M | Flat and raised erosions | Yes | Monoclonal (κ) | None | No | - |
| 27 | Zhang et al. [9] | 28/M | Erythema | No | Monoclonal (κ) | None | No | - |
| 28 | Zhang et al. [9] | 24/F | Erythema | No | Monoclonal (κ) | None | No | - |
| 29 | Zhang et al. [9] | 66/M | Ulceration | No | NA | None | No | - |
| 30 | Joo [48] | 56/M | Hyperemia and micronodularity | Yes | Monoclonal (κ) | MALT lymphoma | No | - |
HPET, Helicobacter pylori eradication therapy; RBG, Russell body gastritis; M, male; F, female; NS, not stated; MGUS, monoclonal gammopathy of undetermined significance; HIV, human immunodeficiency virus; EBV, Epstein-Barr virus; HCV, hepatitis C virus; NA, not assessed; MALT, mucosa-associated lymphoid tissue.
![]()
Immunoglobulin light chain restriction is generally considered to be proof of monoclonality and is an important indicator of B-cell neoplasia. Interestingly, light chain restriction was detected via immunohistochemistry in 12 out of 30 cases of RBG (kappa restriction in 11 cases and lambda restriction in one case) [9,20,48,58,61]. Ten of these cases were localized gastric lesions with no associated lymphoid malignancy or plasma cell disorder, although one case was associated with low-grade MALT lymphoma [48], and another with concomitant monoclonal gammopathy of undetermined significance [20]. This could be explained by the findings of Girón and Shah [63], who reported that approximately 50% of patients with H. pylori infection exhibit either kappa or lambda light chain elevation, and suggested that H. pylori infection might contribute to immunoglobulin light chain dysfunction. Thus, RBG may be closely associated with H. pylori infection, and the majority of RBG cases may be reactive in nature, even when light chain monoclonality is detected. Nevertheless, pathologists should be aware of the possibility of concomitant lymphoid neoplasms.
Given the strong association with H. pylori infection, it is possible that RBG, gastric carcinoma, or MALT lymphoma might occur simultaneously in the same patient. Previous reports describe Mott cell proliferation (features of RBG) in association with gastric carcinoma, including two cases of Epstein-Barr virus–positive lymphoepithelioma-like carcinoma [55], and one case of signet ring cell carcinoma [57]. In these cases, Mott cell proliferation was likely a reactive paraneoplastic event, given that the carcinoma and Mott cells did not mix and that the Mott cells were polyclonal. However, in the previously described case of RBG with concomitant MALT lymphoma [48], the Mott cells were mixed with the neoplastic centrocyte-like cells (Fig. 3C) and exhibited IgM kappa monoclonality, which indicates the proliferating Mott cells were neoplastic components of MALT lymphoma.
Morphologically, Mott cells with eccentric nuclei and abundant eosinophilic cytoplasm are similar to poorly differentiated carcinoma cells or signet ring cells. In addition, in cases with abundant Mott cells and a few neoplastic cells, neoplastic cells may not be easily detected. Thus, immunostaining for cytokeratin should be conducted to exclude associated carcinoma [55,57]. Furthermore, if light chain monoclonality is detected, pathologists should consider associated MALT lymphoma and perform ancillary immunostaining (e.g., for CD20 and cytokeratin) to identify centrocytelike cells and lymphoepithelial lesions (Fig. 3D) [48].
Crystal-storing histiocytosis
CSH is a rare condition, which often occurs with disorders such as monoclonal gammopathy, B-cell lymphoma, or plasma cell myeloma [43,64,65]. Although many cases of CSH are systemic, organ-confined CSH has been described in the lung, lymph node, kidney, thyroid, thymus, parotid gland, and cornea [43,65-71]. CSH is extremely rare in the stomach, and only eight cases of gastric CSH have been described to date in the English literature (Table 2) [10,11,43,71,72]. Among these, H. pylori infection was identified in four patients (50%) who did not exhibit concomitant gastric lesions (except for H. pylori gastritis) or a systemic disorder that might have caused monoclonal gammopathy [10,11]. In the other four patients, there was no mention of H. pylori infection [43,71,72], and two of them were subsequently diagnosed with thymic lymphoma [43] and plasma cell myeloma [71], respectively. Therefore, although overproduction of immunoglobulin due to H. pylori infection could be a plausible cause of isolated gastric CSH, clinical workup is needed to exclude the possibility that it is a manifestation of underlying lymphoma or plasma cell myeloma. Light chain restriction was detected in five of seven cases (kappa restriction in two case and lambda restriction in three cases) [10,71,72], all of which except one had no associated B-cell/plasmacytic neoplasms. Thus, light chain restriction detected in isolated gastric CSH does not necessarily mean that it is associated with B-cell or plasmacytic neoplasm. Meanwhile, because no studies have reported CSH responding to H. pylori eradication therapy, its effectiveness is unclear.
Table 2.
Clinical and pathologic findings of six cases of gastric crystal-storing histiocytosis
| Case No. | Study | Age (yr)/Sex | Endoscopic finding | Ig light chains | Crystal-ctoring cells | Helicobacter pylori infection |
|---|---|---|---|---|---|---|
| 1 | Jones et al. [43] | 35/F | NS | Polyclonal | Histiocytes | NS |
| 2 | Stewart and Spagnolo [10] | 82/M | Gastritis | Monoclonal (IgAλ) | Plasma cells | Positive |
| 3 | Stewart and Spagnolo [10] | 81/M | Gastritis | NA | Plasma cells | Positive |
| 4 | Stewart and Spagnolo [10] | 52/F | Gastritis | Monoclonal (IgAλ) | Plasma cells | Positive |
| 5 | Joo et al. [11] | 56/F | Polyps (three) | Polyclonal | Plasma cells and histiocytes | Positive |
| 6 | Vaid et al. [72] | NS | Submucosal tumor | Monoclonal (κ) | Histiocytes | NS |
![]()
Histologically, CSH is characterized by diffuse infiltrations of large, oval, polygonal, and, occasionally, spindle cells, with abundant eosinophilic cytoplasm and small eccentric nuclei. The eosinophilic cytoplasm is filled with elongated, rectangular, and needle-shaped/fibrillary crystalline inclusions (Fig. 4). These crystalline inclusions are approximately 5–20 nm long and are frequently arranged in parallel arrays [11,43,66]. At low magnification, nodular aggregates of these cells can sometimes resemble adult rhabdomyomas or granular cell tumors in the way that they expand or displace normal structures, and proliferations of benign-looking histiocyte-like cells can also resemble Gaucher disease or malakoplakia [43,65,73,74]. Therefore, immunostaining for desmin, smooth muscle actin, S100 protein, and immunoglobulin light chains can facilitate an accurate diagnosis [11,43,65,66,74].
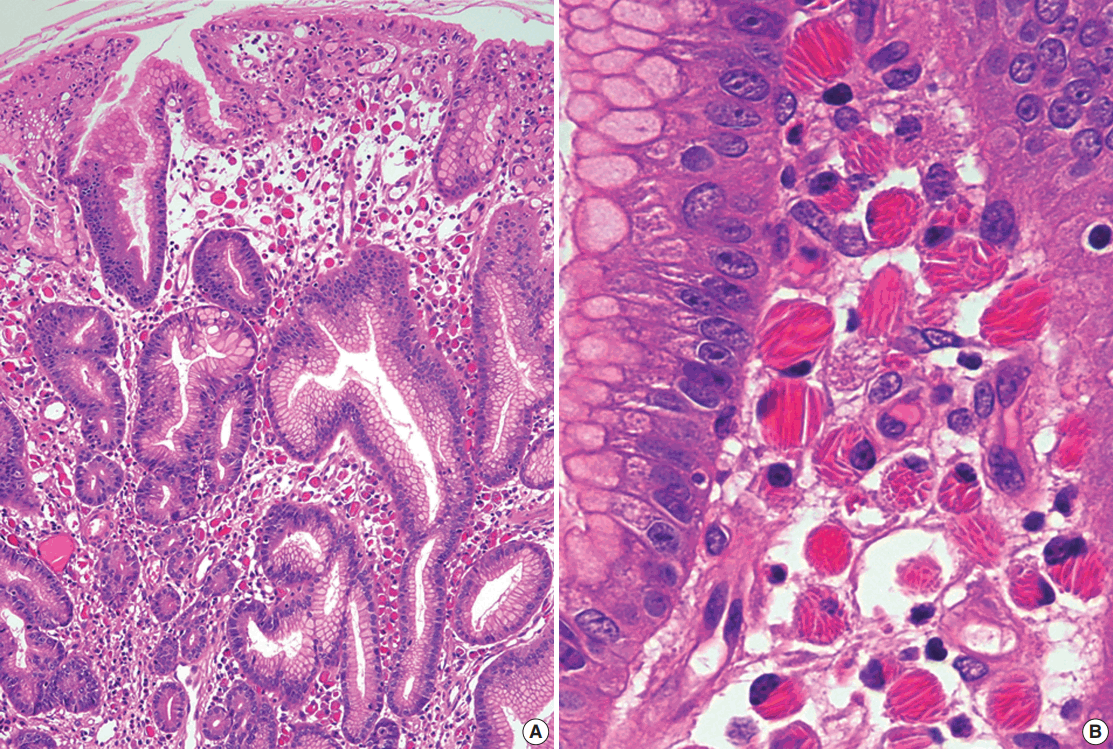
Go to :
CONCLUSION
This review examined several rare gastric lesions that have distinctive histological characteristics and are associated with a variety of conditions, including H. pylori infection. Although H. pylori may be a cause in many of these conditions, the association cannot be viewed as definite, given the low incidence of these lesions relative to the high prevalence of H. pylori infection, regional differences in the prevalence of H. pylori infection, and the possibility of other causative disorders. However, it is reasonable to consider H. pylori once other potential etiologies have been excluded. In addition, it is not advisable to consider H. pylori to be an “innocent bystander,” given the considerable proportion of these lesions that can be regressed or cured with H. pylori eradication therapy. Therefore, it is important that pathologists properly identify a lesion’s cause in order to ensure appropriate patient management. In this context, H. pylori should be considered as a possible cause in areas where it is prevalent. Further investigation is needed to confirm the role of H. pylori in the development of rare gastric lesions.
Go to :
 XML Download
XML Download