

 PDF
PDF Citation
Citation Print
Print


Inflammatory myofibroblastic tumor is a histopathologically distinctive neoplasm that has been described in various anatomic sites of children and young adults. It is composed of myofibroblastic spindle cells admixed with infiltrates of plasma cells, lymphocytes and eosinophils [1]. Most common site is the lung followed by other sites. In 1954, Umiker and Iverson [2] first described it in the lung. According to World Health Organization (WHO) classification, inflammatory myofibroblastic tumor is an intermediate tumor (rarely metastasizing), with potential for recurrence. These tumors were initially considered as reactive in nature, however, the neoplastic nature was revealed by molecular analysis that showed clonal rearrangement of the anaplastic lymphoma kinase (ALK) gene on the short arm of chromosome 2 at 2p23 with fusion of the 3' kinase region of the ALK gene with various partners including TPM3, TPM4, CLTC, and ranbinding protein 2 (RANBP2) in 50%–70% of inflammatory myofibroblastic tumors [3].
A few cases of inflammatory myofibroblastic tumors undergo malignant transformation displaying large polygonal cells and higher mitotic rate including atypical mitosis [4]. Marino-Enriquez et al. [4] in 2011 first described the term epithelioid inflammatory myofibroblastic sarcoma because of the malignant nature of these tumours. Inflammatory myofibroblastic tumors expressing RANBP2 and ALK rearrangement (RANBP2-ALK) fusion usually display high grade epithelioid/round cell morphology with nuclear membrane staining pattern and predict poor prognosis [5].
Expression of ALK fusion proteins in inflammatory myofibroblastic tumors can be detected by immunohistochemistry (IHC), the staining pattern of which is determined by the fusion partner: diffuse cytoplasmic staining when the partnering proteins are TPM3, TPM4, CARS, ATIC, and SEC31L122; granular cytoplasmic staining with CLTC; and nuclear membrane staining with RANBP2 [4].
However, the biological nature may be aggressive even when the histologic appearance is bland [6]. There were no definite histopathologic, molecular, or cytogenetic features to predict the malignant transformation, recurrence, or metastasis [7]. Surgical resection is the preferred treatment for inflammatory myofibroblastic tumors. Nonsteroidal anti-inflammatory drugs (NSAIDs), steroids, chemotherapy, and radiotherapy have been used as adjuvant therapy when feasible [8].
We undertook this retrospective study to correlate the histological atypia, multifocal appearance of tumor (multicentric in origin), recurrence, and metastasis with the immunohistochemical expression of ALK-1 in inflammatory myofibroblastic tumors, so as to objectively predict the biological behavior.
MATERIALS AND METHODS
This was a retrospective study of 18 patients with inflammatory myofibroblastic tumors, diagnosed in the Department of General Pathology during the period of January 2011–December 2015. The study was approved by the institutional review board (IRB No. 10200). The clinical details and follow-up information of each patient were noted from the clinical work station. All the archived slides were taken out and reviewed. Wherever necessary, blocks were cut and fresh slides prepared. All classical tumors fulfilled the morphologic criteria described in the WHO Classification of Tumours of Soft Tissue and Bone [3]. Atypical cases were defined by increased cellularity, cellular atypia with large ganglion-like round to polygonal cells, multinucleated or anaplastic giant cells, atypical mitosis and necrosis [7]. The archival tissues obtained from institutional and consultation files were fixed in 10% buffered neutral formalin and paraffin-embedded tissue blocks were prepared. Hematoxylin and eosin (H&E) stained sections were made. IHC was done on representative paraffin block(s) from each tumor.
IHC procedure
IHC slides and H&E slides prepared from formalin fixed, paraffin embedded tissue blocks were reviewed. Fresh sections were cut from archival blocks when necessary. IHCs had been done on Ventana Benchmark XT (Ventana Medical Systems, Tucson, AZ, USA). Details of primary antibodies used for IHC are shown in Table 1. Appropriate positive controls were used for each antibody throughout the study. The stain was considered to be positive if the tumor cells showed specific cytoplasmic and/or nuclear membrane staining for the particular antibody. The immunohistochemical stains were evaluated semiquantitatively as follows: 0, negative; 1+, < 10% of cells positive; 2+, 10%–50% of cells positive; and 3+, > 50% of cells positive [7].
Intra-abdominal tumors with epithelioid morphology, nuclear or perinuclear accentuation of ALK immunopositivity and aggressive clinical behavior were categorized as epithelioid inflammatory myofibroblastic sarcoma [4].
RESULTS
A total of 18 cases of inflammatory myofibroblastic tumors were found during the study period. Of the 18 cases, 17 were institutional cases and one was a consultation case (case 2) from outside hospital. The clinicopathological features and outcome in the study group are depicted in Table 2. There were nine females and nine males with female to male ratio of 1:1 and the mean age was 23.8 years (range, 3 to 44 years). The most common locations were lung (six cases) and intra-abdominal region (six cases) followed by suprarenal, mastoid antrum, dorsum of nose, mediastinum, sphenoid wing, and breast (one case each). All the patients underwent surgical excision and a few patients had combination treatments: surgery plus chemotherapy or chemoradiotherapy. Limited follow-up information was available, ranging from 3 to 65 months (mean, 16.1 months). One patient had died of disease. Two cases were lost for follow-up. Remaining patients were alive with no evidence of disease at last follow-up.
Pathological findings
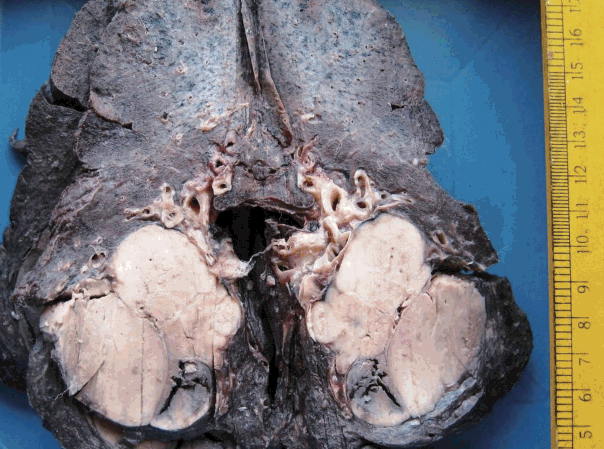
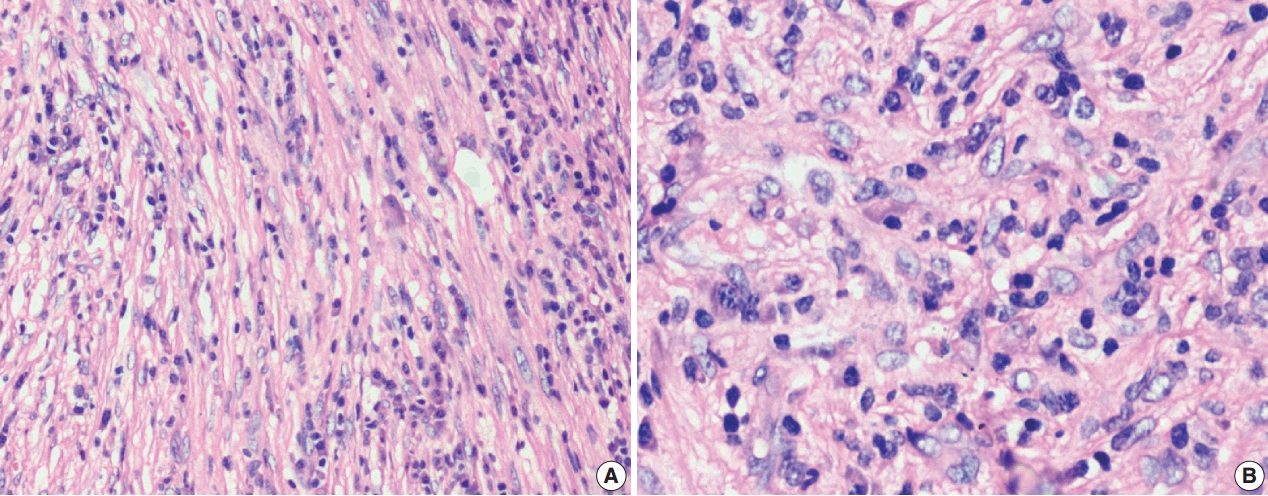
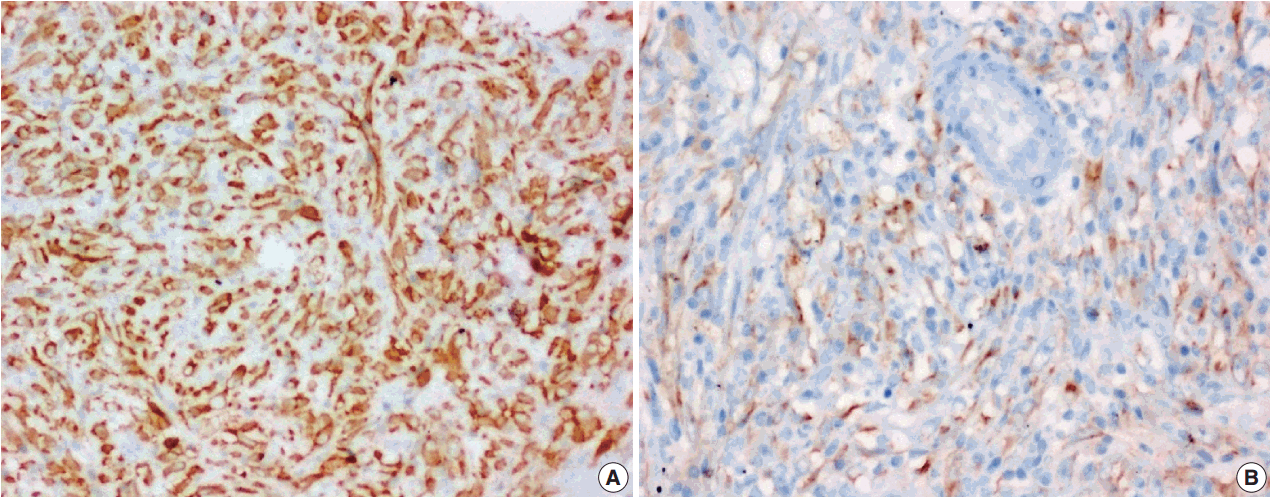
The tumor size ranged from 1 to 15 cm (mean, 5.6 cm). Grossly, the tumors were nodular circumscribed masses (Fig. 1) exhibiting soft to firm variegated appearance with grey white to tan fleshy and focal mucoid areas. One case showed variable hemorrhage and necrosis on the cut surface (case 1). Three cases (cases 13, 15, and 18) had multifocal appearance of tumor (multicentric in origin) in various anatomic locations (Table 2). Histologically, among the 18 inflammatory myofibroblastic tumors (including recurrent and metastatic cases), 14 cases had classic histologic features (Fig. 2) and four cases had atypical histologic features, revealing diffuse and focal cytoplasmic IHC expression of ALK-1 (Fig. 3).
Classification of histological patterns was done for all the cases according to the dominant tumor area, which included cellular (77.8%), myxoid (11.1%), and fibrous (11.1%) types. All the classical inflammatory myofibroblastic tumors displayed a bland spindle cell proliferation admixed with an inflammatory infiltrate composed of lymphocytes, plasma cells, and eosinophils. The atypical histologic features included increased cellularity, cellular atypia with large ganglion-like round to polygonal cells, multinucleated or anaplastic giant cells, atypical mitosis, and necrosis.
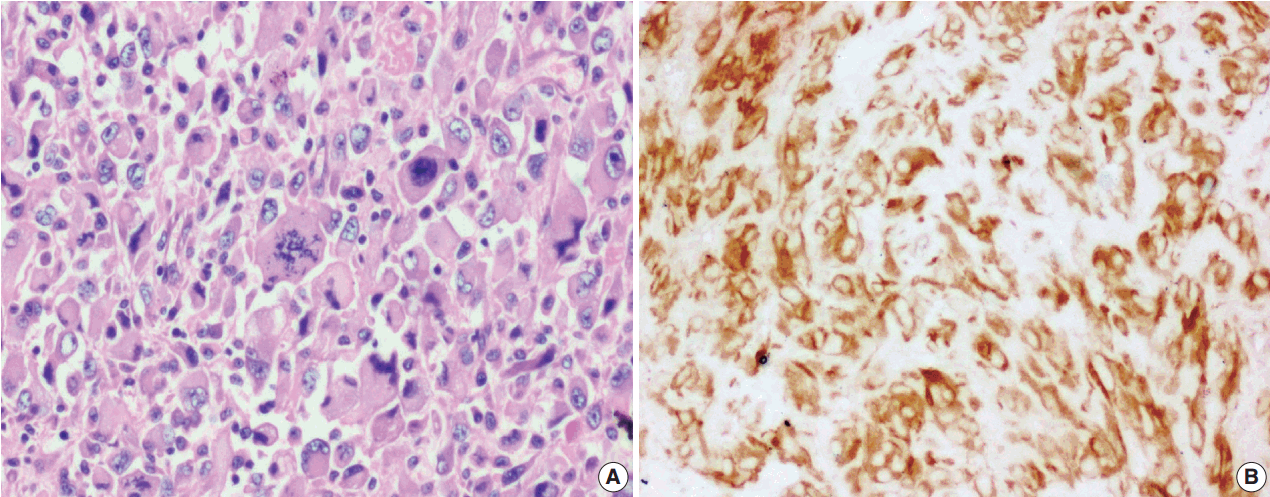
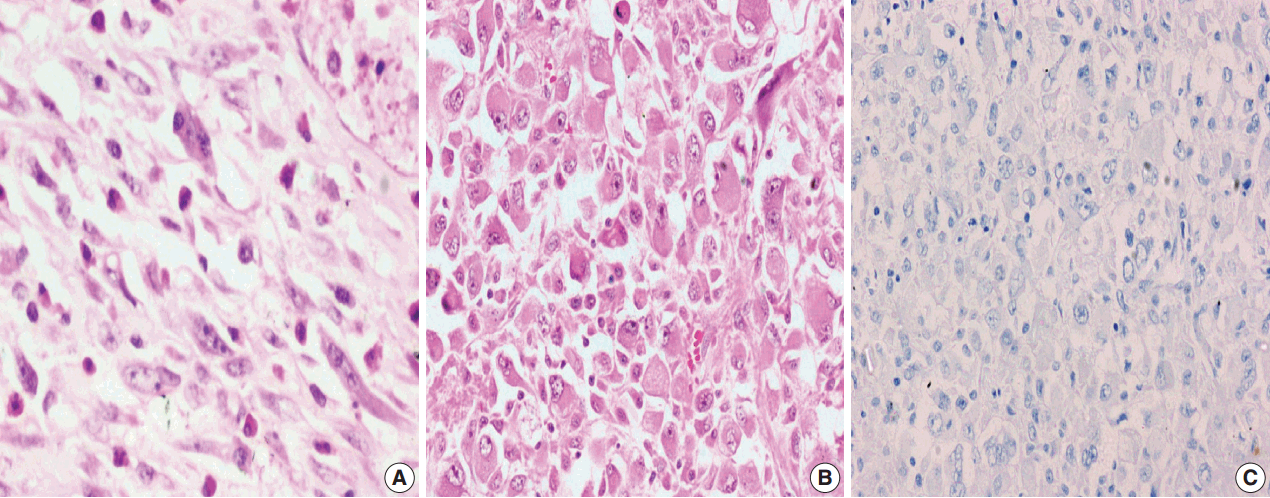
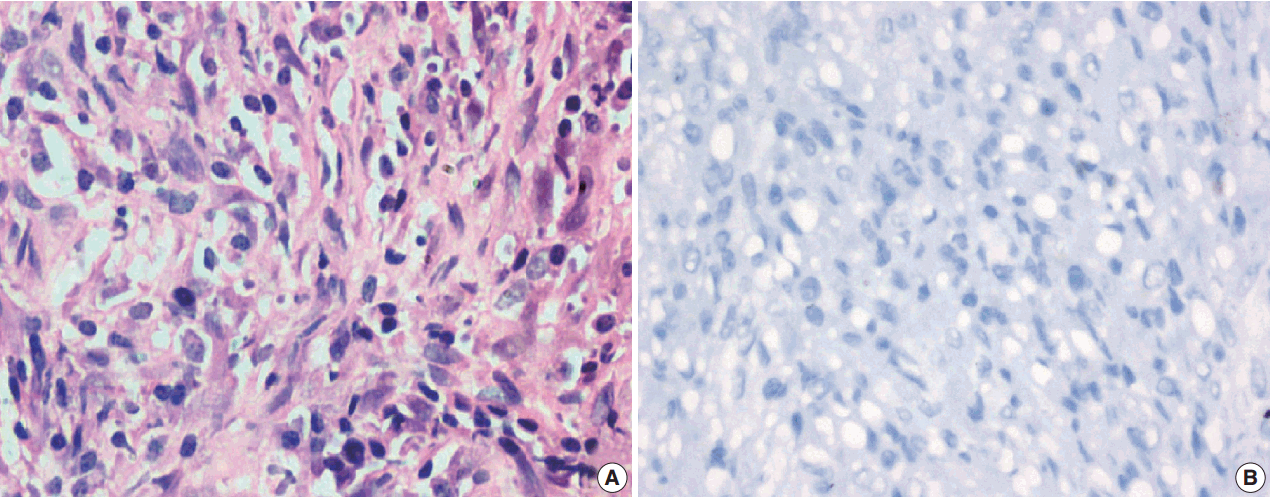
Among the atypical cases, one case (case 1) showed dominant epithelioid morphology with focally ganglion like cells, hypercellularity, atypia, mitosis, and prominent nucleoli (Fig. 4); one case (case 13) showed dominant spindle cell morphology and focal epithelioid morphology (Fig. 5); remaining two cases (cases 2 and 6) showed atypical spindle cell morphology (Fig. 6).
Among the atypical cases, three cases had recurrence (cases 1, 6, and 13) and two cases had metastasis to the lung, liver (case 1), and pelvic bone (case 15).
Immunohistochemical findings
Spindled tumor cells of inflammatory myofibroblastic tumors showed cytoplasmic or membrane positivity for smooth muscle actin (12/16 cases), muscle-specific actin (3/6 cases), vimentin (8/8 cases), desmin (5/11 cases), and epithelial membrane antigen (2/4 cases). The spindled tumor cells were negative for cytokeratin, myogenin, CD34, CD21, CD117, h-caldesmon, TLE-1, CD30, and CD15 (Table 3).
Immunohistochemical stain for ALK-1 expression was available in all the 18 cases, demonstrating cytoplasmic reactivity of spindled or polygonal tumor cells for ALK-1 in 10 cases (55.6%) and no reactivity in eight cases (44.4%) (Tables 3, 4). Diffuse (3+) cytoplasmic staining was observed in six cases and focal (2+) cytoplasmic staining in three cases.
An intra-abdominal inflammatory myofibroblastic tumor with dominant epithelioid morphology (case 1) revealed perinuclear accentuation of ALK-1 immunoexpression, suggestive of epithelioid inflammatory myofibroblastic sarcoma (Fig. 4); the remaining atypical cases with spindle cell morphology (including case 13 with focal epithelioid morphology) were negative for ALK-1, suggestive of atypical inflammatory myofibroblastic tumors only (Figs. 5, 6).
ALK-1 positive tumors (55.6%) included five females and five males and the mean age was 23.8 years (range, 3 to 44 years), compared with the mean age of (four females and four males) 27.1 years (range, 18 to 41 years) for ALK-1 negative cases. Recurrence was noted in 3/10 ALK-1 positive (30%) and 3/8 ALK-1 negative (37.5%) cases, whereas metastasis to the lung, liver and pelvic bone was noted in the ALK-1 positive group (Tables 4, 5).
Statistical analysis
ALK-1 immunopositivity in neither of the classical (p = .275), atypical (p = .275) nor multifocal tumor (p > .99) types was statistically significant at 5% level of significance with Fisher exact test. There was no statistically significant correlation between ALK-1 expression and either of the recurrence (p > .99) or metastasis (p = .485) of inflammatory myofibroblastic tumors.
DISCUSSION
Inflammatory myofibroblastic tumors formerly described as inflammatory pseudotumor, with an unpredictable behavior ranging from benign to low-grade malignant, are classified as intermediate grade tumors, with potential for recurrence and rare metastasis, in the current WHO Classification of Tumours of Soft tissue and Bone [3]. These tumors are described in children and young adults in various anatomic sites with a tendency towards local recurrence in about 25% of abdominopelvic lesions and rare metastasis (< 5%) [3,7]. A few cases of inflammatory myofibroblastic tumors may appear morphologically benign but the biological nature may be aggressive even when the histologic appearance is bland. There were no definite predictors of the histologic transformation, recurrence, or metastasis. These tumors were considered as a neoplasm after the discovery of clonal rearrangement of the ALK gene on the short arm of chromosome 2 at 2p23 in 50%–75% of extrapulmonary inflammatory myofibroblastic tumors [7].
In a study by Wang et al. [9], the most common site of inflammatory myofibroblastic tumors was abdominopelvic region (73.9%) followed by other locations. The present study included 33.3% each of intrapulmonary and intra-abdominal locations followed by other locations. There was slight male predominance in the literature [10,11]; however, there was an equal incidence in each gender in the present study.
Hussong et al. [12] in 1999 stated that the combination of cellular atypia, presence of ganglionlike cells, p53 expression and DNA aneuploidy are all possible predictors to identify the malignant change and aggressive behavior of inflammatory myofibroblastic tumors. In a study by Coffin et al. [7], 27.1% were classical and 72.9% were atypical inflammatory myofibroblastic tumors. The atypical features in his study were increased cellularity, cellular and nuclear pleomorphism, distinct fascicular and focal herringbone pattern, cellular atypia with large ganglion-like round to polygonal cells, multinucleated or anaplastic giant cells, atypical mitosis, and necrosis. ALK immunopositivity was noted in 78% atypical, 54% recurrent and 60% classical inflammatory myofibroblastic tumors. Metastatic inflammatory myofibroblastic tumors (10.1%) were negative for ALK expression.
In another study [13], there were 60% classical and 40% atypical inflammatory myofibroblastic tumors. Twenty-six point seven percent cases showed ALK expression. Twenty-five percent of ALK-positive and 63.6% of ALK-negative inflammatory myofibroblastic tumors revealed recurrence. Metastasis was restricted to the ALK-negative group. Statistically significant correlation was found between atypia, recurrence and metastasis. There was a significant correlation between ALK expression and recurrence. The present study revealed 77.8% classical and 22.2% atypical inflammatory myofibroblastic tumors. Overall ALK-1 immunopositivity was noted in 55.6% and negativity in 44.4% of inflammatory myofibroblastic tumors. Metastatic inflammatory myofibroblastic tumors were ALK-1 positive in the present study. There was no statistically significant correlation between ALK-1 expression, atypia, recurrence and metastasis in the present study compared to the other studies, probably due to a small sample size and limited follow up data; a larger study may be taken up in the future to evaluate the statistical significance.
Marino-Enriquez et al. [4] proposed a term epithelioid inflammatory myofibroblastic sarcoma for epithelioid variant of intraabdominal inflammatory myofibroblastic tumors with a nuclear membrane or perinuclear pattern of ALK expression and aggressive clinical behavior. In the present study, one patient with an intra abdominal mass, which was categorized as inflammatory myofibroblastic sarcoma, with epithelioid morphology and perinuclear accentuation of ALK-1 expression, developed liver and lung metastasis and expired within 8 months. ALK gene rearrangement was demonstrated in all the tested cases of epithelioid variant of inflammatory myofibroblastic tumors by fluorescence in situ hybridization (FISH) [4]. Janik et al. [14] studied pulmonary and extrapulmonary pseudotumors with overall recurrence rate of 14%. Multifocality of pulmonary and extrapulmonary pseudotumors had a high chance of recurrence compared with tumors confined to a single organ. Intraabdominal pseudotumors were associated with > 75% of the recurrences when compared to other extrapulmonary pseudotumors. In the present study, extrapulmonary multifocal inflammatory myofibroblastic tumors were associated with recurrence and metastasis. Study by Chaudhary [15] pointed out that ALK gene rearrangements were often seen in inflammatory myofibroblastic tumors of children and young adults compared to adults over 40 years of age. The search for ALK gene rearrangement by FISH is a useful tool for the diagnosis of inflammatory myofibroblastic tumors.
Ramotar et al. [16] reported a case of inflammatory pseudotumor of the head and neck mimicking a metastatic disease in a human immunodeficiency virus–positive 49-year-old female. In immunocompromised patients, the likelihood of inflammatory pseudotumor should also be considered in the differential diagnosis. ALK protein immunoexpression was detected in various other tumors including lipomatous tumors, rhabdomyosarcomas, Ewing’s sarcoma/primitive neuroectodermal tumors, malignant fibrous histiocytomas, and leiomyosarcomas. Except inflammatory myofibroblastic tumors, almost all other tumors displayed low level of ALK expression [17].
The differential diagnosis of these tumors includes both benign and malignant spindle or epithelioid cell tumors. The spindle cell tumors include fibroblastic/myofibroblastic tumors, fibrous histiocytoma, and solitary fibrous tumor [6]. The tumors with epithelioid or round cell morphology include anaplastic large cell lymphoma, epithelioid leiomyosarcoma, rhabdomyosarcoma, undifferentiated sarcoma, myxoid/round cell liposarcoma, myxofibrosarcoma, dedifferentiated liposarcoma, poorly differentiated carcinoma, malignant melanoma, and epithelioid gastrointestinal stromal tumor [4,5].
Anaplastic large cell lymphoma can be differentiated from epithelioid variant of inflammatory myofibroblastic sarcoma by immunoreactivity for CD30, CD3, and CD43, diffuse membranous or cytoplasmic staining of ALK-1 and absence of RANBP2-ALK rearrangement. High-grade leiomyosarcoma usually reveals fascicles of spindle and epithelioid cells with increased cellularity, cellular atypia, pleomorphism, cigar-shaped nuclei and abundant brightly eosinophilic cytoplasm. The tumor cells are positive for h-caldesmon and negative for ALK staining. Alveolar rhabdomyosarcoma shows uniform round cells with scant cytoplasm and usually lacks myxoid background and inflammatory infiltrates. Immunohistochemical stain for myogenin reveals nuclear positivity. Dedifferentiated liposarcoma can have features of inflammatory myofibroblastic tumor, but with expression of MDM2 and absence of ALK-1. Epithelioid malignant peripheral nerve sheath tumor will be strongly and diffusely positive for S100 and negative for ALK-1 and also SMARCB1. Malignant melanoma shows polygonal cells with prominent nucleoli and the cells are positive for S100 and human melanoma black 45. Epithelioid gastrointestinal stromal tumors reveal immunoexpression of CD117, discovered on GIST-1 (DOG1) and negative for ALK-1 [3-5].
Surgical resection is the preferred treatment for inflammatory myofibroblastic tumors. NSAIDs, steroids, chemotherapy, and radiotherapy have been used as adjuvant therapy when feasible [8].
In a study by Jacob et al. [18], systemic (liver and lumbar spine) involvement of inflammatory myofibroblastic tumor in a 45-year-old female, expressing ALK gene rearrangement, responded very well to ALK inhibitor (Crizotinib). In a series of cases by Butrynski et al. [19], multifocal intra-abdominal inflammatory myofibroblastic tumors which expressed ALK gene rearrangement, were treated successfully by ALK inhibitor (Crizotinib). Fragoso et al. [11] in 2011 stated that, although surgical resection is the preferred treatment for inflammatory myofibroblastic tumors, cases which are not amenable to surgical excision may be treated conservatively.
In conclusion, this study demonstrates that inflammatory myofibroblastic tumors exhibit a wide age group distribution with an equal incidence in males and females. Most of the tumors with histological atypia were associated with recurrence, irrespective of ALK-1 expression. Epithelioid inflammatory myofibroblastic sarcoma with perinuclear accentuation of ALK-1 showed aggressive clinical course with metastasis and mortality. Few cases which were benign in histology also exhibited recurrence irrespective of ALK-1 expression. Metastasis was seen in ALK-1 positive tumors. However, there was no statistically significant correlation between ALK-1 expression, tumor type, recurrence, and metastasis. Overall, ALK-1 IHC is a useful diagnostic aid in the appropriate histomorphologic context, especially in identifying epithelioid inflammatory myofibroblastic sarcomas. There were no definite histopathologic criteria to predict malignant transformation, recurrence, or metastasis. However, this study is limited by the very small number of cases, limited follow-up data and also the lack of identification of ALK gene rearrangement by FISH. A search for newer molecular tools by larger studies needs to be performed to identify prognostic features to guide treatment and predict the outcome.
 XML Download
XML Download