

 PDF
PDF Citation
Citation Print
Print


An extrapleural solitary fibrous tumor (SFT) is an uncommon mesenchymal spindle cell tumor characterized by its unpredictable behavior [1,2]. It can occur in any site of the body [3]. Approximately 10% of SFTs are found to be locally aggressive and may present local or distant recurrence many years after primary treatment [4]. The distinction between benign and malignant SFT (MSFT) is difficult and does not have established specific criteria in the current literature. According to the World Health Organization classification of soft tissue tumors, MSFT is designated as a tumor that has features of hypercellularity, at least focal moderate to marked cellular atypia, tumor necrosis, 4 mitoses/10 high-power fields (HPFs), and infiltrative margins [5].
Heterologous differentiation of MSFT is extremely rare and has been described in only one article with osteosarcomatous and rhabdomyosarcomatous elements [6]. We herein present another case of MSFT with heterologous rhabdomyosarcomatous differentiation.
CASE REPORT
A 56-year-old man presented with a newly-developed large mass in his left posterior thigh. He had been treated 6 months earlier with palliative chemoradiation for unclassified sarcomas of the cervical spine, the right femoral head, and both lungs. Magnetic resonance imaging revealed a poorly marginated, 8.5-cm, heterogeneously enhancing mass in the left thigh with nodular, heterogeneous bone marrow signal intensity in the right femur neck, in the proximal femoral shaft, and in the pelvic bone, suggesting intramedullary bone metastasis. Wide excision of the thigh mass was performed under the assumption of high-grade sarcoma. The resected mass showed a multinodular, dark yellow, fish-flesh appearance with necrotic foci measuring 10.6×6.4×2.3 cm (Fig. 1).
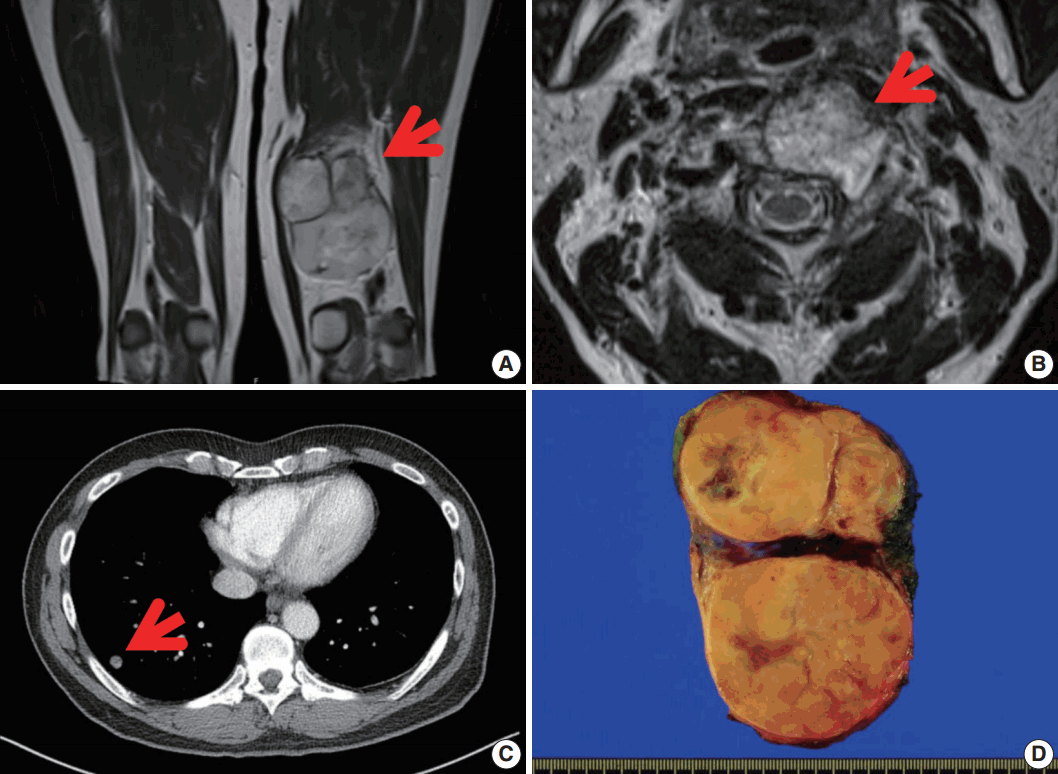 | Fig. 1.Magnetic resonance image revealing a well-defined enhancing mass in the postero-medial compartment of the left thigh (arrow) (A), with metastasis in the C2 vertebral body (arrow) (B). (C) Chest computed tomography showing multiple nodules in both lungs. (D) Grossly, a large lobulated mass is observed in the left thigh. The cut surface of the mass is yellow-tan and firm with necrotic foci.
|
Microscopically, the tumor showed heterogeneous cellularity with partly cystic changes and a hemangiopericytic vascular pattern. A hypercellular area showed spindle cells or epithelioid cells with high mitotic activity (63/10 HPFs), whereas a hypocellular area and cystic wall showed pleomorphic rhabdoid cells (Fig. 2). The hypercellular area was immunopositive for CD34 (1:500, Immunotech, Marseille, France) and CD99 (1:200, Dako, Glostrup, Denmark), whereas the hypocellular area and cystic wall showed immunoreactivity for desmin (1:200, Dako) and myogenin (1:200, Dako, Carpinteria, CA, USA) (Fig. 3). The patient received adjuvant chemoradiation therapy (pazopanib 2 cycles and 3,000 cGy radiation 7 cycles) and had shown no recurrence after 7 months of follow-up.
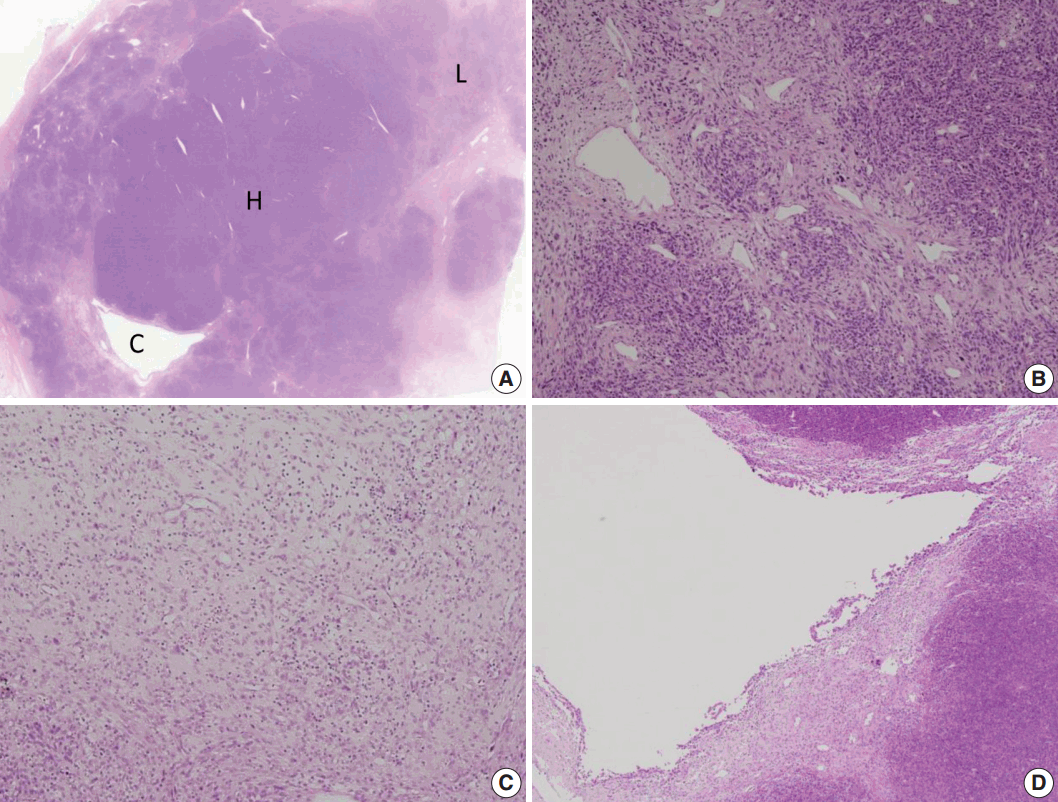 | Fig. 2.Microscopic findings. (A) The mass consists of a hypercellular area (H), a hypocellular area (L), and a cystic area (C). (B) The hypercellular area is composed of short spindle or epithelioid cells with a hemangiopericytic vasculature. (C) The hypocellular area shows plump epithelioid cells with inflammatory cells. (D) The cystic area is unilocular and contacted both the hypercellular and the hypocellular areas.
|
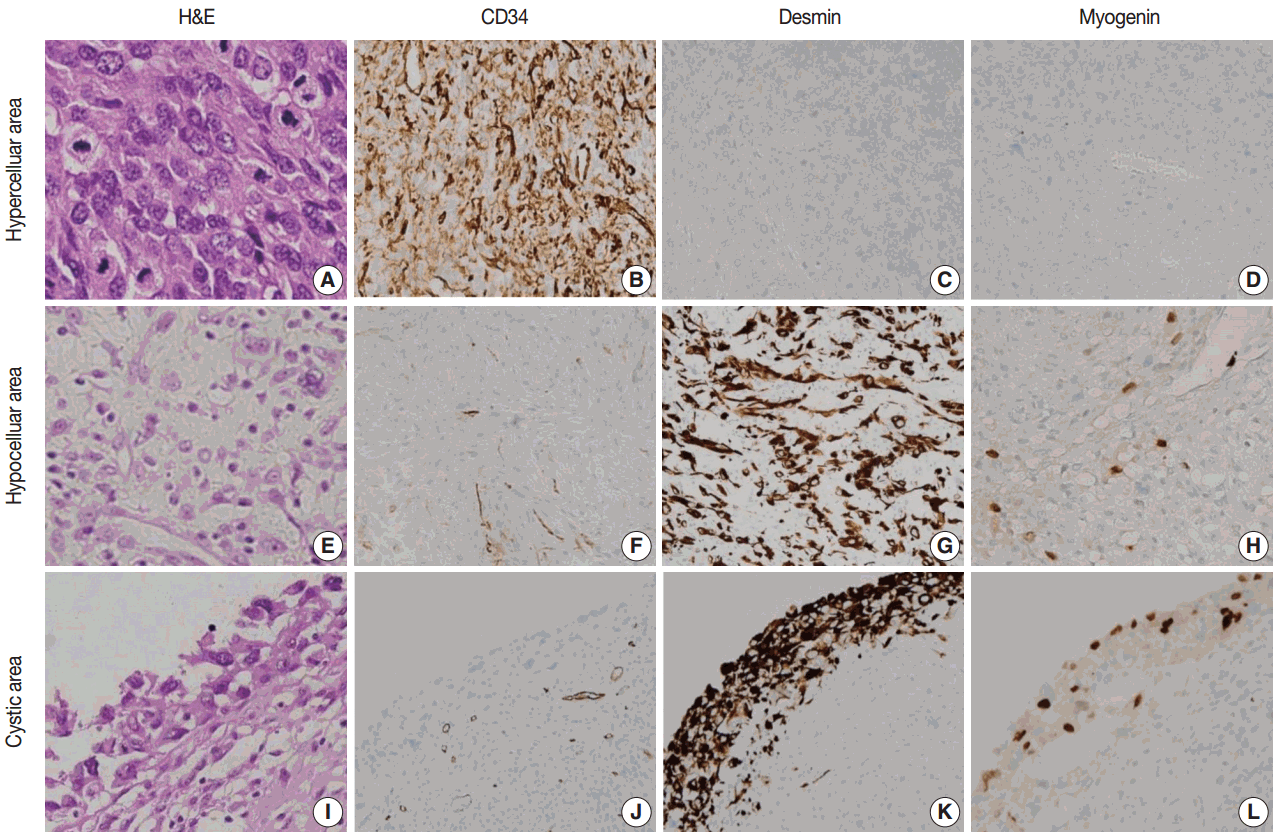 | Fig. 3.Morphologic and immunohistochemical findings. (A) The hypercellular area is composed of short spindle or epithelioid cells with prominent nucleoli and high mitotic activity (63/10 highpower fields) and shows immunoreactivity for CD34 (B) and immunonegativity for desmin (C) and myogenin (D). (E) The hypocellular area is composed of plump epithelioid cells and shows immunoreactivity for CD34 (F) and strong immunopositivity for desmin (G) and myogenin (H). (I) The cystic component is composed of pleomorphic rhabdoid cells and shows focal immunoreactivity for CD34 (J) and strong immunopositivity for desmin (K) and myogenin (L).
|
Go to : 
DISCUSSION
SFT is a mesenchymal tumor that is characteristically ‘‘patternless’’ with an arborescent vascular frame [5]. It can occur at any site of body and can vary in appearance, including hyaline, cellular, and epithelioid elements. It presents as well-demarcate and slow growing [7]. A malignant form of SFT is rare, and it has been proposed to arise either de novo or within a preexisting benign SFT [8]. There are some reported cases of MSFT arising from benign SFT. Yokoi et al. [8] previously described three cases of malignant transformation from benign SFT. The tumors had both a malignant-looking and a benign-looking component. The malignant-looking component had highly atypical cells with hyperchromatic nuclei and increased mitotic activity and had markedly high expression of p53, Ki-67, and CD34 and immunonegativity for desmin and myogenin, whereas the benign-looking component included low- or intermediate-grade tumors with typical SFT features. Another five cases of de novo MSFT have also been described [8]. These cases had heterogeneous, high- to low-grade pathologic features.
As previously mentioned, only one case of MSFT with heterologous components has been described to date [6]. This patient was a 59-year-old male with a 10-cm-sized mass in the medial aspect of the thigh who underwent a wide excision. The tumor had three distinct characteristics. First, it was a typical SFT with strong immunopositivity for CD34 and Bcl-2 and low mitotic activity (2/10 HPFs). Second, it consisted of pleomorphic cells with nuclear atypia and many bizarre multinucleate tumor giant cells with immunonegativity for CD34 and a high mitotic activity (23/10 HPFs). In addition, this area was also admixed with desmin- and myogenin-immunopositive rhabdomyosarcomatous elements. Third, the tumor showed osteosarcomatous differentiation with distinct malignant osteoid cells. After wide excision, that patient proceeded to postoperative radiotherapy and remained free of disease for 7 months.
In our case, the patient was a 56-year-old man that had a 10.6-cm-sized, newly developed large mass in his left posterior thigh. He had been treated with palliative chemoradiation 6 months earlier for unclassified sarcomas of multiple metastasis. Microscopically, the tumor showed three different components: hypercellular, hypocellular, and partly cystic components. Cystic components presented with a hemangiopericytic vascular pattern. A hypercellular area showed spindle cells or epithelioid cells with high mitotic activity and showed immunopositivity for CD34 and CD99, whereas a hypocellular area and cystic wall showed pleomorphic rhabdoid cells and immunoreactivity for desmin and myogenin. The patient received adjuvant chemoradiation therapy and has shown no recurrence after 7 months of follow-up.
The differential diagnosis of MSFT includes benign and malignant lesions, such as malignant peripheral nerve sheath tumor (MPNST), synovial sarcoma (SS), fibrosarcoma, undifferentiated pleomorphic sarcoma (previously malignant fibrous histiocytoma), and dermatofibrosarcoma protuberans (DFSP) [5,9]. MPNST has features of heterogeneous spindle cells with variable growth pattern and arrangement and has bizarre giant cells, high mitotic activities, and distinguishing patterns of necrosis [10]. MPNST also shows focal immunopositivity for S100 protein.
SS has three major histological subtypes: biphasic, monophasic, and poorly differentiated. The biphasic subtype shows co-existence of epithelioid cells and spindle cells. However, the monophasic subtype is entirely composed of spindle cells; in this case, immunonegativity for CD34 is helpful to exclude the diagnosis [11].
Fibrosarcoma consists of highly cellular fibroblasts with variable collagen production. It has a herringbone-like growth pattern and consists of scant cytoplasm, hyperchromatic nuclei, and variable nucleoli [5]. It also presents immunonegativity for CD34.
Undifferentiated pleomorphic sarcoma has high cellularity lesions, mixed with spindle cells and often rounded histiocyte-like cells. Some cases have extensive fibrous stroma. This tumor has a storiform growth pattern and pleomorphic tumor cells with foamy cytoplasm and marked nuclear atypia. Multinucleated giant cells may also be seen.
DFSP presents with a non-circumscribed, highly cellular lesion and has a storiform growth pattern. Cells are monomorphic and spindly with scant eosinophilic cytoplasm. It usually has no significant pleomorphism and rare histiocytes [5]. DFSP is less deeply located than SFT and usually involves the dermis [12].
Barthelmess et al. [13] identified recurrent somatic fusions of two genes, NGFI-A binding protein 2 (NAB2) and STAT6, located at 12q13, as presumable tumor-initiating events in SFT. Although the authors raised the possibility that specific NAB2-STAT6 fusion variants may be associated with higher risk of aggressive behavior [13], there are no distinct molecular features that differentiate benign from malignant tumors.
Surgical excision has been the standard treatment option for both benign and MSFTs, but late recurrences have been observed [14]. Radiotherapy is often used to improve local control, and chemotherapy is used for lesion that cannot be completely excised [15]. However, the benefits of these adjuvant chemoradiation therapies remain unproven. In conclusion, we present a rare case of MSFT with rhabdomyosarcomatous differentiation. This case report expands our knowledge of the histologic features of MSFT.
Go to :
 XML Download
XML Download