

 PDF
PDF Citation
Citation Print
Print


Genetic alterations in non-small cell lung cancer (NSCLC) have been identified in recent years [1-4]. Epidermal growth factor receptor (EGFR), v-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS), and anaplastic lymphoma kinase (ALK) are the most commonly mutated oncogenes that involve the pathogenesis of lung cancer as “genetic drivers.” Advanced NSCLC has a dismal prognosis with a short median overall survival [5], but several selective EGFR tyrosine kinase inhibitors (TKIs) and an ALK inhibitor show effectiveness as personalized target therapy in patients who harbor those specific genetic mutations [3,6]. According to guidelines from the College of American Pathologists, the International Association for the Study of Lung Cancer (IASLC) and the Association for Molecular Pathology, mutation analysis is recommended and recent studies have demonstrated the cost-effectiveness of genetic screening, especially in Asians, who have a higher prevalence of EGFR mutations [7,8].
Chromosomal alterations, closely related to unique histologic phenotypes through protein expression, determine differentiation, patterns, and cell types. The identification of typical morphological features of certain mutations allows retrospective speculation on underlying genetic alterations. Although EGFR, KRAS, and ALK mutations have generally been considered mutually exclusive in NSCLCs [9], some cases harbor double mutations in a single tumor [10-13]. Identifying driver oncogenes in double-mutated tumors through morphologic differences and comparing the responsiveness of targeted therapies can help explain the pathogenesis of lung cancer and predict resistance to targeted therapy. Hence, we explored histologic features and genetic drivers of tumors with concomitant mutations and discovered the dominant mutation in carcinogenesis for each.
MATERIALS AND METHODS
A retrospective review was performed of biopsied and/or surgically resected cases diagnosed as NSCLC and tested for EGFR, KRAS, and ALK mutations in the Department of Pathology and Translational Genomics at Samsung Medical Center, Seoul, Korea from 2006 to 2014. Of 6,637 NSCLC patients, 6,595 EGFR, 5,177 KRAS, and 4,869 ALK mutation tests were performed. Among them, 2,387 patients (36.2%) had EGFR, 398 (7.7%) had KRAS, and 281 (5.8%) had ALK mutations. Based on these tests, we selected 12 patients with concomitant mutations in the same tumor.
Clinical data, including gender, age, smoking history, and previous concurrent chemoradiation therapy history were extracted from electronic medical records. All hematoxylin and eosin stained slides of selected cases were reviewed by two pathologists (T.L. and B.L.). Histologic classification was made according to the IASLC/American Thoracic Society (ATS)/European Respiratory Society (ERS) International Multidisciplinary Classification of Lung Adenocarcinoma, and typical pathologic features were re-evaluated, including cell type, nuclear and cytoplasmic characteristics.
We used two methods to evaluate EGFR mutations in the 18th, 19th, 20th, and 21st exon: Sanger sequencing and real time polymerase chain reaction after peptide nucleic acid (PNA)–clamping using the PNA clamping EGFR Mutation Detection Kit (Panagene, Inc., Daejeon, Korea). KRAS mutations were evaluated with Sanger sequencing in the 12th and 13th exon. Extracted genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) tissue was used for EGFR and KRAS analyses. ALK mutation tests were performed using immunohistochemical (IHC) (1:40, NCL-ALK, clone 5A4, Novocastra, Newcastle upon Tyne, UK) and fluorescence in situ hybridization (FISH) analysis (LSI ALK dual color break-apart probe, Dako, Glostrup, Denmark) with FFPE tissue. Diffuse strong positivity in the cytoplasm was regarded as positive in ALK IHC. In the FISH analysis, 50 non-overlapping nuclei were counted and 15% of break-apart probes were used as a cutoff value for positivity. Diffuse strong positive ALK IHC results were interpreted as a surrogate marker of ALK rearrangement [14].
Go to : 
RESULTS
Clinical characteristics
Clinical data and pathologic features of the 12 patients with concomitant mutations are summarized in Table 1. The age at diagnosis of patients ranged from 48 to 78 years old (median, 62 years old) and 75% of patients were female. All three male patients had a history of smoking (15–57 pack-years) in contrast to none of female patients. Five surgically resectable patients underwent lobectomy and seven clinical stage IV patients had a needle aspiration or biopsy from the lung, bronchus, lymph node, or adrenal gland.
Table 1.
Clinicopathologic features of 12 patients with double mutated pulmonary adenocarcinoma
| Patient No. | Sex/Age(yr) | Smoking | Specimen | Stage | EGFR mutation | Additional mutation | Dominant pattern | Typical cell feature | Mucin | Intranuclear inclusion | Surgery | Targeted therapy | Response | Status | PFS (mo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/74 | 57PY | Lung/R | pT1bN1 | E21 L858R | KRAS I21S | Papillary and micropapillary | Hobnail cells | No | No | + | - | - | DODa | - |
| 2 | F/62 | Never | Lung/A | M1(IV) | E21 L858R | KRAS G12D | b | No | No | Present | Gefitinib | PR → PD | DOD | 7 | |
| 3 | F/63 | Never | Lung/R | pT1bNO | E21 L858R | KRAS G13A | Papillary and acinar | Hobnail cells | No | No | + | - | - | NED | - |
| 4 | F/48 | Never | Lung/R | pT1bN2 | E19 deletion | KRAS G12V | Acinar and solid | Hobnail and columnar cells | Intra- and extracytoplasmic | No | + | Erlotinib | PR → PD | DOD | 20 |
| 5 | M/55 | 20PY | Lung/R | pT2N2 | E19 deletion | KRAS G13C | Solid and acinar | Hobnail cells | No | Present | + | Gefitinib | PR | AWD | 29 |
| 6 | F/78 | Never | Lung/B | M1(IV) | E18 G719X | KRAS G12D | Papillary and acinar | Hobnail cells | No | No | - | (refused) | - | AWDa | - |
| 7 | F/62 | Never | LN/B | M1(IV) | E21 L858Rc | ALK | Solid and cribriform | Signet ring cells | Intracytoplasmic | No | +d | Gefitinib | PR | AWDa | 18e |
| 8 | F/62 | Never | Lung/B | M1(IV) | E21 L858R | ALK | Solid and cribriform | No | No | No | - | Gefitinib and Crizotinib | SD and PR | AWD | 24 |
| E18 G719X | |||||||||||||||
| 9 | M/56 | 15PY | Lung/B | M1(IV) | E18 G719Xc | ALK | Solid | No | No | No | Crizotinib | SD | DOD | 4 | |
| 10 | F/68 | Never | Lung/R | ypT2N2 | E18 G719Xc | ALK | Solid, micropapillary and cribriform | Signet ring cells | Intra- and extracytoplasmic | Present | + | - | - | NED | - |
| 11 | F/58 | Never | Lung/B | M1(IV) | E19 deletion | ALK | Solid | Signet ring cells | Intracytoplasmic | No | - | Crizotinib | - | AWDa | e |
| 12 | F/66 | Never | Adrenal/B | M1(IV) | E20 R803W | ALK | Solid | No | No | No | +d | Erlotinib | PD | AWDa | 0.7 |
EGFR, epidermal growth factor receptor; PFS, progression-free survival; M, male; PY, pack-year; R, resection; E, exon; KRAS, v-Ki-ras2 Kirsten rat sarcoma viral oncogene; DOD, died of disease; F, female; A, aspiration; PR, partial response; PD, progressive disease; NED, no evidence of disease; AWD, alive with disease; B, biopsy; LN, lymph node; ALK, anaplastic lymphoma kinase; SD, stable disease.
![]()
Patient No. 5 received EGFR TKI targeted therapy for bone metastasis at 15 months after lobectomy. Patient No. 6 refused treatment with chemo- or targeted therapy and patient No. 7 had a history of lobectomy at an outside hospital without any other treatment 9 years prior to the lymph node biopsy. Patient No. 10 received neoadjuvant concurrent chemoradiation therapy prior to surgery. Patient No. 12 previously underwent lobectomy and was treated with gefitinib for 1 month and crizotinib for 4 months in China. She had no EGFR or KRAS mutation at the time of diagnosis at China. Aside from this patient, all other patients had no history of prior targeted therapy before mutation testing.
Mutational and histologic characteristics
All 12 patients were diagnosed with adenocarcinoma and had an EGFR mutation. EGFR mutations of three patients were only detected with the PNA clamping method, while nine were confirmed by Sanger sequencing. Five (41.7%) had the missense L858R mutation in exon 21, four (33.3%) had the missense G719X mutation in exon 18, and three (25%) had a deletion in exon 19. Patient No. 8 had the L858R and G719X mutations at the same time and patient No. 12 had a missense R803W mutation at exon 20 that was not identified before targeted therapy at the outside hospital. No patient had a T790M point mutation, which is associated with resistance. Six patients had additional KRAS mutations and the other six had an ALK mutation (referred to as ‘EGFR-KRAS’ and ‘EGFR-ALK’ hereafter). Three among the six patients who showed positivity at ALK IHC were confirmed with FISH analysis.
EGFR-KRAS and EGFR-ALK tumors showed distinct morphologic characteristics. Six EGFR-KRAS patients had papillary, acinar, solid, and micropapillary patterns (Fig. 1). Among them, five patients had typical hobnail cell features with apical snouts; the remaining patient (patient No. 2) underwent needle aspiration and did not show any typical cell features. One patient (patient No. 4) had a focal columnar cell component and intraand extracytoplasmic mucin, which are similar morphologic features of NSCLC with a KRAS mutation. The other five patients did not show any mucin. Two of the six patients had intranuclear inclusions.
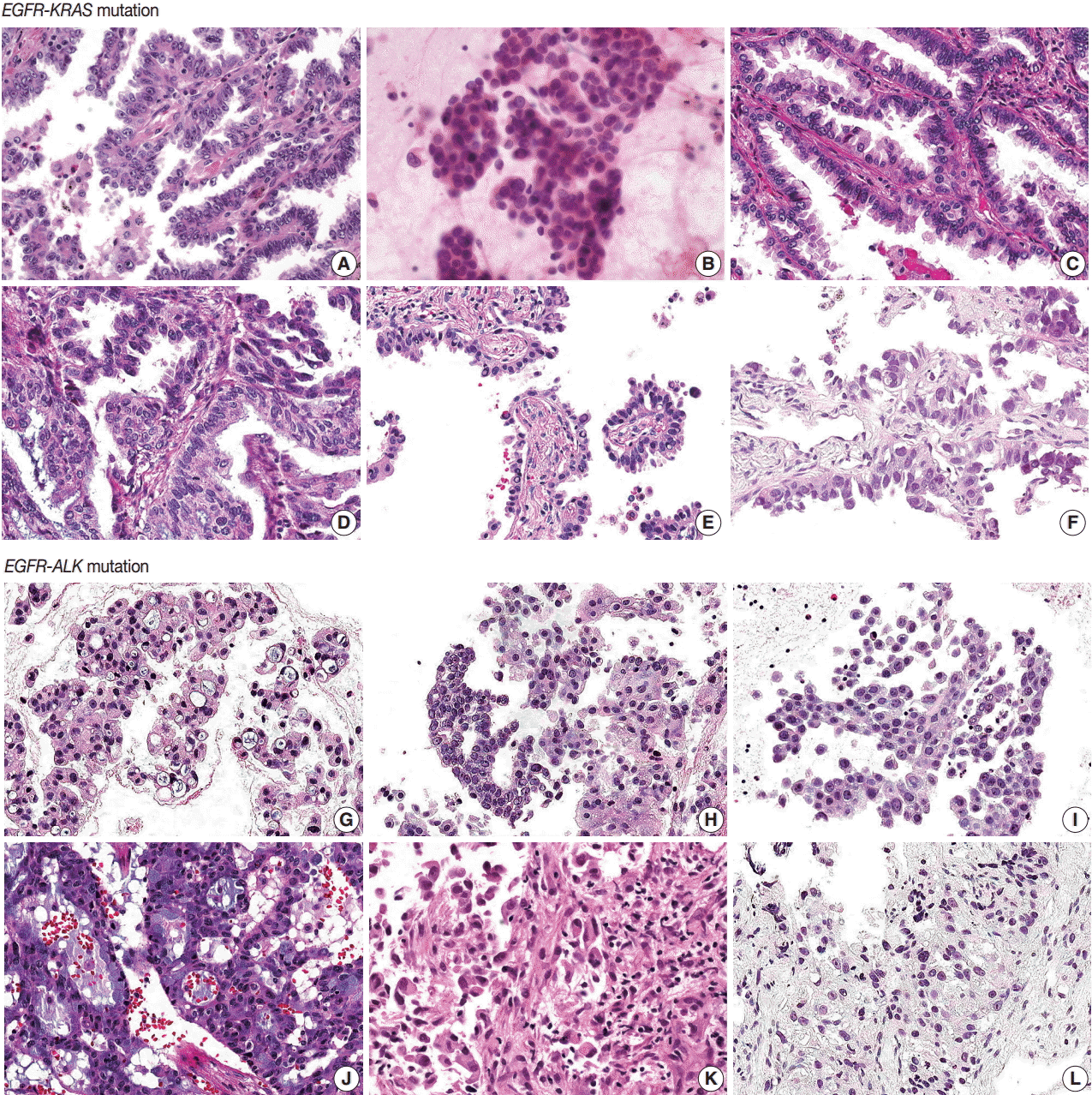 | Fig. 1.Histologic features of 12 pulmonary adenocarcinomas with concomitant mutations. (A–F) In the six EGFR-KRAS patients, patients No. 1 (A), No. 3 (C), No. 5 (E), and No. 6 (F) have papillary, micropapillary and acinar patterns with hobnail cells. (D) Patient No. 4 has an acinar pattern and hobnail cells for the most part but shows focal columnar cells with intra- and extracellular mucin. (B) Patient No. 2 does not show any typical cell features. (G–L) In the six EGFR-ALK patients, all patients show solid, cribriform or micropapillary patterns rather than the papillary or acinar patterns that are easily identified as EGFR-KRAS tumors. Patients No. 7 (G), No. 10 (J), and No. 11 (K) have signet ring cells with intra- or extracytoplasmic mucin. But in the other three patients, No. 8 (H), No. 9 (I), and No. 12 (L), typical cell features are not identified, as neither signet ring cells nor hobnail cells. EGFR, epidermal growth factor receptor; KRAS, v-Ki-ras2 Kirsten rat sarcoma viral oncogene; ALK, anaplastic lymphoma kinase.
|
In contrast, six EGFR-ALK patients showed solid, cribriform and micropapillary patterns. Three of six patients had signet ring cells and intracytoplasmic mucin with or without extracytoplasmic mucin production, which are typical features of ALK-positive NSCLC. The others did not show any typical cell features or mucins. One patient had intranuclear inclusions.
Treatment responses and follow-up status
Seven patients had surgical resection and two were alive without targeted therapy and any evidence of relapse or progression, including one patient who received neoadjuvant concurrent chemoradiation therapy. One patient who had surgery and decided not to receive targeted therapy due to old age and underlying diabetes mellitus died of disease after 53 months from surgery (patient No. 1).
EGFR TKI included gefitinib (250 mg, orally, every day) and erlotinib (150 mg, orally, every day) and the ALK inhibitor was crizotinib (250 mg, orally, every day or twice daily). Three among six EGFR-KRAS patients received targeted therapy with EGFR TKI. All three patients showed partial responses, but two also showed disease progression after 7 and 20 months. In the EGFR-ALK patients, two were treated with EGFR TKI, two with ALK inhibitor and one received both EGFR TKI and ALK inhibitor therapy. Most patients treated with ALK inhibitor or EGFR TKI showed partial response or stable disease, but patient No. 12 with EGFR TKI showed progressive disease.
Go to :
DISCUSSION
Concomitant genetic alteration of NSCLC is unusual because EGFR, KRAS, and ALK mutations are widely known as mutually exclusive. Most are associated with acquired mutation after targeted therapy and are related to drug resistance. In previous studies, two different hypotheses were proposed to explain dual mutation in NSCLC [12]. Underlying intratumoral heterogeneity of EGFR mutations [15], different tumor cells may have different oncogenic driver mutations. Some authors have also suggested coexistence of mutations in the same tumor cells using IHC expression and mutation-specific antibodies [12,16].
EGFR, as a growth factor membrane-bound receptor tyrosine kinase, controls cell proliferation and survival. Approximately 36% of patients with NSCLC in East Asia have EGFR mutations and 90% of mutations are exon 21 L858R or exon 19 deletion [17]. These mutations increase tyrosine kinase activity, resulting in hyperactivation of the RAS-RAF-MEK-ERK pathway, which regulates cell proliferation, and the phosphoinositide 3-kinase–AKT–mammalian target of rapamycin pathway, which is associated with cell survival. RAS proteins are central mediators in EGFR tyrosine kinase signaling and also associated with cell proliferation and differentiation. In particular, KRAS mutations are common in lung cancer but less common in East Asia than in Western countries [18]. ALK is a receptor tyrosine kinase. ALK fusion with various N-terminal fusion partners stimulates kinase activity, and signaling of these mutations are involved in cell growth, proliferation and apoptosis. Among NSCLCs, 3%–7% harbor ALK fusion [3,4] and the majority of these fusions are associated with the echinoderm microtubule-associated proteinlike 4 (EML4) gene. EML4-ALK fusions are associated with EGFR TKI resistance [19].
Typical histologic features of lung cancer with EGFR, KRAS, and ALK mutations have been described [20-24]. All of these mutations are frequent in adenocarcinoma and EGFR mutations are more common in tumors with papillary, micropapillary, acinar, and lepidic patterns than in others. Hobnail cell type and intranuclear inclusion are typical nuclear features of EGFR mutation-harboring tumors. A solid pattern and mucinous type are more frequent in KRAS mutation tumors. Extracellular mucin production, cribriform patterns, and signet ring cell features are more common in ALK mutation tumors.
In this study, we analyzed six EGFR-KRAS (1.5%) and six EGFR-ALK patients (2.1%) among KRAS and ALK mutation-positive NSCLC patients. The majority of EGFR-KRAS tumors showed typical histologic features of EGFR mutation, except one case which had focal morphologic features similar to KRAS mutation. In a previous study, NSCLC harboring a KRAS mutation showed resistance to EGFR TKI and adjuvant chemotherapy [2]. In our study, however, three patients showed partial response to gefitinib and erlotinib for 7–29 months. Intratumoral heterogeneity of the mutation may explain this discordance between coexistent KRAS mutation and response to EGFR TKI therapy. Patient No. 2 had short progression-free survival (PFS) and disease progressed rapidly, while the other two patients had 20–29 months of PFS. Based on the evidence of morphologic phenotypes and responsiveness to targeted therapy, we expected these two patients to have coexistent mutations in the same tumor cells, and hypothesized that the EGFR mutation was the dominant driver oncogene in lung cancer, even if the tumor cells harbored an additional KRAS mutation.
In EGFR-ALK patients, some previous studies have shown acquired EGFR mutations as a mechanism of resistance to ALK inhibitor therapy [25,26]. In our case, patient No. 12 had no EGFR or KRAS mutation and was treated with crizotinib in an outside hospital. After 4 months of ALK-targeted therapy, she acquired an additional EGFR mutation and showed poor response to erlotinib, which is consistent with previous studies. In the other four patients, however, diverse responses were identified with no resistance to gefitinib or crizotinib. Conflicting results of responsiveness to EGFR TKI and ALK inhibitor in EGFR-ALK patients in NSCLC have been reported in a few limited studies [10,27]. Recently, Yang et al. [16] investigated relationships between receptor phosphorylation and response to targeted therapy, and suggested that relative phospho-EGFR and ALK levels can be used to predict response.
Although the G719X mutation accounts for about 7% of EGFR mutated NSCLC in East Asia [17], three of five EGFR-ALK patients had the EGFR exon 18 G719X missense mutation in our study. In addition, three of five EGFR-ALK patients showed typical histologic features of ALK-expressing adenocarcinoma. From these findings, we suspect that the ALK mutation determines morphological phenotypes and acts as a driver oncogene, and the EGFR mutation may also play an important role in oncogenesis. Accordingly, based on evaluation of the signaling pathway, including phosphorylation of receptor kinase, the modification or combination of EGFR TKI and ALK inhibitor offer promising treatments.
The small number of cases limited our analysis. Baldi et al. [12] revealed the possibility of more frequent concomitant mutations than expected and Won et al. [13] reported an increased proportion of NSCLC harboring concomitant EGFR and ALK mutations from 4.4% to 15.4% using more sensitive methods such as targeted next-generation sequencing (NGS) and mutant-enriched NGS. Greater numbers of previously undisclosed concomitantly mutated tumors can be identified using more sensitive and advanced techniques. Some authors also suggest that tumor burden of each mutation affects responses to targeted therapy. There may be a limitation to evaluating mutations from biopsy specimens, but the majority of patients who need to receive targeted therapy might be clinically unresectable and may only be diagnosed with biopsy. Thus, the expectation of tumor burden in a limited biopsy specimen is unfavorable.
To the best of our knowledge, this is the first study to describe double mutated NSCLC with histologic findings. Morphologic findings might help predict underlying driver mutations and further studies are warranted.
In conclusion, we investigated genetic driver mutations in 12 double mutated NSCLCs with analysis of clinical and pathologic features. The majority of EGFR-KRAS tumors showed typical histologic patterns and cell features of EGFR-mutated tumors and partially responded to EGFR TKI. In EGFR-ALK tumors, however, some showed ALK-mutated features and partially responded to ALK inhibitor or EGFR TKI. EGFR and ALK play an important role in the oncogenesis of NSCLC and tumor morphology can provide important clues to predict treatment response. We suggest that patients with histologic features of EGFR mutations can be treated with EGFR TKI, even with coexistence of a KRAS mutation. We also suggest that EGFR-ALK patients should have underlying mutational pathways evaluated and be treated with a selection and/or combination of ALK inhibitor and EGFR TKI. Further studies are needed to support these interesting findings.
Go to :
 XML Download
XML Download