

 PDF
PDF Citation
Citation Print
Print


This tumor was originally recognized in patients who underwent surgery for treatment of medically intractable seizures; however, recent progress in neuro-imaging has allowed for increased detection of dysembryoplastic neuroepithelial tumors (DNTs) in patients with a single episode of epilepsy or in older patients [1]. The original report in 1988 by Daumas-Duport et al. [2] describes morphologically unique features, including intracortical multi-nodularity, a specific glioneuronal (GN) element, and association with focal cortical dysplasia (FCD). The term “dysembryoplastic neuroepithelial tumor” was introduced for this unique tumor because tumors have a number of clinicopathological features, strongly suggesting a dysembryoplastic origin such as early onset of seizure, presence of FCD in the adjacent cortex, and deformity of the overlying skull [2]. The generic term “neuroepithelial tumors” was used so that a large range of morphologic variants could be integrated into this entity [2].
The complex type of DNTs was first categorized as neuronal and mixed neuronal-glial tumors in the 1993 World Health Organization (WHO) classification of central nervous system (CNS) tumors. Additionally, simple and nonspecific histologic variants of DNTs have been described. The simple types of DNTs consist of only specific GN elements and the complex types are characterized by specific GN elements associated with glial nodules and/or FCD. Nonspecific types of DNTs lack multinodular architecture and specific GN elements, but show similar histologic findings to those seen in glial nodules of complex DNTs [3,4]. Nonspecific DNTs have been controversial because their histology is indistinguishable from conventional gliomas and because of the lack of a specific GN element and/or multinodularity of typical DNTs. The 2007 WHO classification of CNS tumors includes the simple and complex subtypes of DNT [5]. However, several studies demonstrated three histologic forms of DNTs under the different terms “multinodular, solitary nodular, and diffuse” or “simple, complex, and diffuse [6-8].” The frequent immunoreactivity of CD34, nestin, and microtubule-associated protein 2 (MAP2) [9] and the identification of BRAFV600E mutation in nonspecific DNTs [10] suggest that DNTs exhibit a broad spectrum of histopathology from simple to nonspecific forms.
A large series of DNTs demonstrated intrinsic epileptogenicity of DNTs in three histologic subtypes by intralesional recordings and simple and complex types showed localization of the epileptogenicity to the tumor, but non-specific temporal DNTs showed more extensive areas with the epileptogenicity. In their series, FCD was found in approximately two-thirds of patients with DNTs and was more common in nonspecific (85%) than in complex types (47%) [11].
CLINICAL FEATURES
DNTs are found in approximately 17.8%–20% of patients who undergo surgical resection for chronic epilepsy [11,12]. DNTs are the second most common tumors in surgically resected cases for intractable epileptic seizures [13,14]. Gangliogliomas and DNTs account for 65% of 1,551 tumors collected at the European Epilepsy Brain Bank [13]. DNTs comprised 87% of 31 cases with tumor-associated, temporal lobe epilepsy [15]. In children (below 18 years of age), the frequency of DNTs was reported to be 0.6% among 340 primary CNS tumors [16]. In another report, the estimated prevalence of DNTs was 0.8% in 233 children with hemato-oncologic problems [17].
Partial complex seizures are the most common clinical manifestation, followed by generalized tonic-clonic, simple partial, and partial seizures with secondary generalization [18]. Secondary generalized seizures are more common in simple DNT cases [6]. An additional neurologic symptom is headache only. Over 90% of patients with DNTs have epilepsy before the age of 20 years [11]. In one of the largest series, the mean age at seizure onset and surgery was 14.6 years (range, 3 months to 54 years), and 30.5 years (range, 6 to 65 years), respectively, with no significant difference among the DNT types [6]. In pediatric DNTs, the mean age at seizure onset and surgery was 8.1 years (2 months–14 years) and 12.4 years (3.25–18.5 years), respectively [19]. Patients generally do not have neurological deficits or evidence of elevated intracranial pressure. Males are more frequently affected, and tumors have a predilection for the temporal lobe, followed by the frontal and parietal or occipital lobes. Multifocal DNTs affecting the different sites in the CNS have been reported [20] and unusual locations for DNTs include the septum pellucidum, caudate nucleus, thalamus, pons, cerebellum, brainstem, and ventricles [21-23]. Familial occurrence of DNTs has been described [24].
Lesionectomy alone controls seizures and tumors in most cases [25]. Seizure recurrence is associated with the presence of residual tumors or FCD in the peritumoral cortex. Therefore, complete removal of tumors and FCD in the adjacent cortex achieves seizure-free outcomes [18]. Chassoux et al. [26,27] reported that favorable prognostic factors for seizure-free outcomes are complete removal of tumor and epileptogenic zones, shorter epilepsy duration, and absence of cortico-subcortical damage at the resection site. The surgical outcome is not different in the three histological types of DNTs. Rarely, tumor recurrence has been reported in cases with gross total or subtotal resection. The histology of recurrent tumors is found to be similar to those of primary tumors. Malignant transformation into high-grade astrocytoma is also reported and may be induced by radiation therapy [6,28]. Aggressive histopathological findings such as increased mitosis, necrosis, microvascular proliferation or high Ki-67 labeling index, and incomplete removal of tumors have not correlated with poor outcomes.
Go to : 
NEUROIMAGING
Cortical topography and the lack of mass effect and perilesional edema are common characteristics of neuroimaging [29]. In a large series of DNT, cystic change (23.9%), expansion of the overlying calvarium (19.5%), focal enhancement (19.5%), and calcification (15.2%) are common radiologic features [6]. On noncontrast computed tomography scans, tumors are hypodense with a cystic appearance in half of the cases and can appear as a calcific hyperdensity and frequently show focal contrast enhancement. Deformity can also be seen in the overlying calvarium that is indicative of chronic focal pressure. Magnetic resonance imaging (MRI) shows a well-delineated tumor with pseudocystic or multicystic appearance, and low signal intensity on T1-weighted images but high signal intensity on T2-weighted images. Focal ring enhancement can rarely be seen on contrast-enhanced T1-weighted images.
Recently, MRI features in histologic variants of DNTs are classified into three types as follows: type 1 (cystic/polycystic-like, well-delineated, strongly hypointense on T1), type 2 (nodularlike, heterogeneous signal), or type 3 (dysplastic-like, isosignal/hyposignal T1, poor delineation, gray-white matter blurring) [29] (Fig. 1A–F). Simple or complex DNTs are always seen as type 1 on MRI, whereas nonspecific DNTs are seen as either type 2 or type 3 on MRI. Epileptogenic zones are found to be significantly different among MRI subtypes. The epileptogenic zone co-localizes to the tumor in type 1 MRI and involves the peritumoral cortex in type 2 MRI, while there are more extensive areas involved in type 3 MRI.
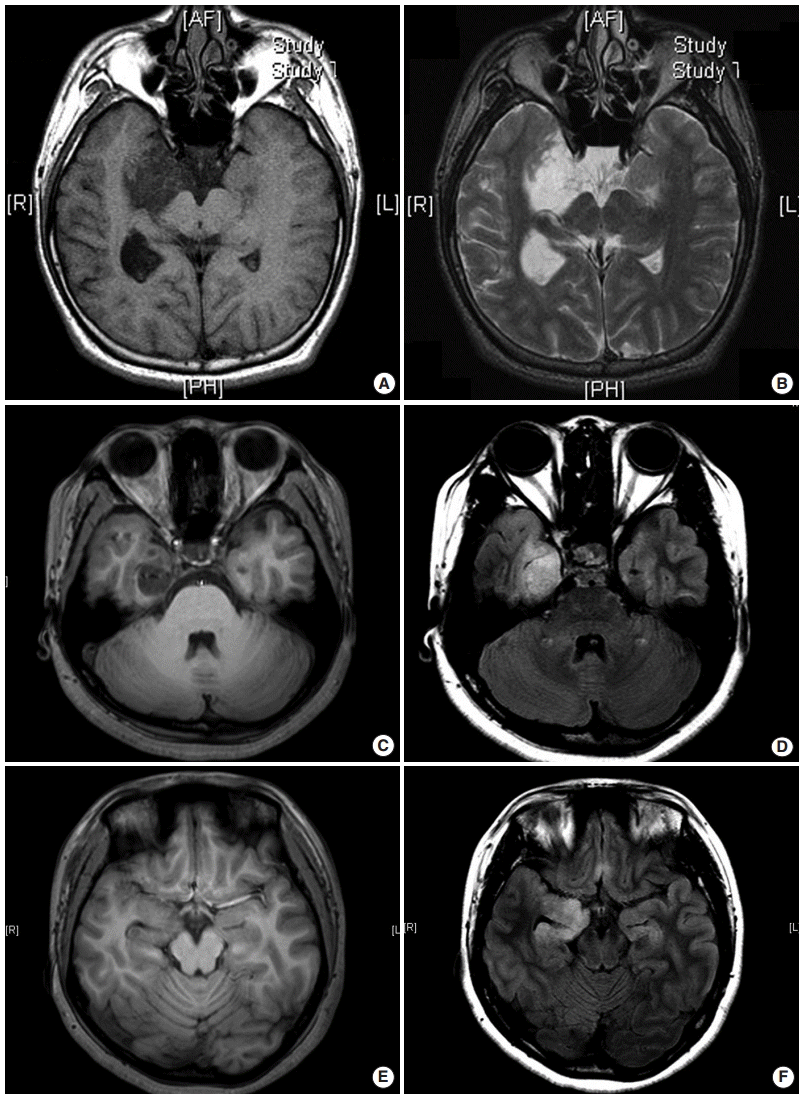 | Fig. 1.Three different types of magnetic resonance imaging in dysembryoplastic neuroepithelial tumors. (A, B) Type 1 shows a well-delineated, polycystic-like tumor with strongly hypointense on T1- and hyperintense on T2-weighted images. (C, D) Type 2 shows a nodular-like, heterogeneous lesion. (E, F) Type 3 shows a poorly delineated, dysplastic-like, iso/hyposignal T1 with gray-white matter blurring. Type 1 is mainly found in simple or complex forms, and type 2 and 3 are observed in nonspecific forms. (A, C, E) T1-weighted images. (B, D, F) T2 FLARE images.
|
Go to :
HISTOPATHOLOGY
Lesions vary in size from 10 to 25 mm, although occasionally larger tumors of up to 70 mm have been reported [11]. Grossly, tumors appear as well-defined, solitary nodular masses or poorly demarcated lesions (Fig. 2A). On the cut section, most tumors are cortically located and may extend into the underlying subcortical white matter in larger tumors. Multi-nodular appearance or cystic changes are commonly found (Fig. 2B, C).
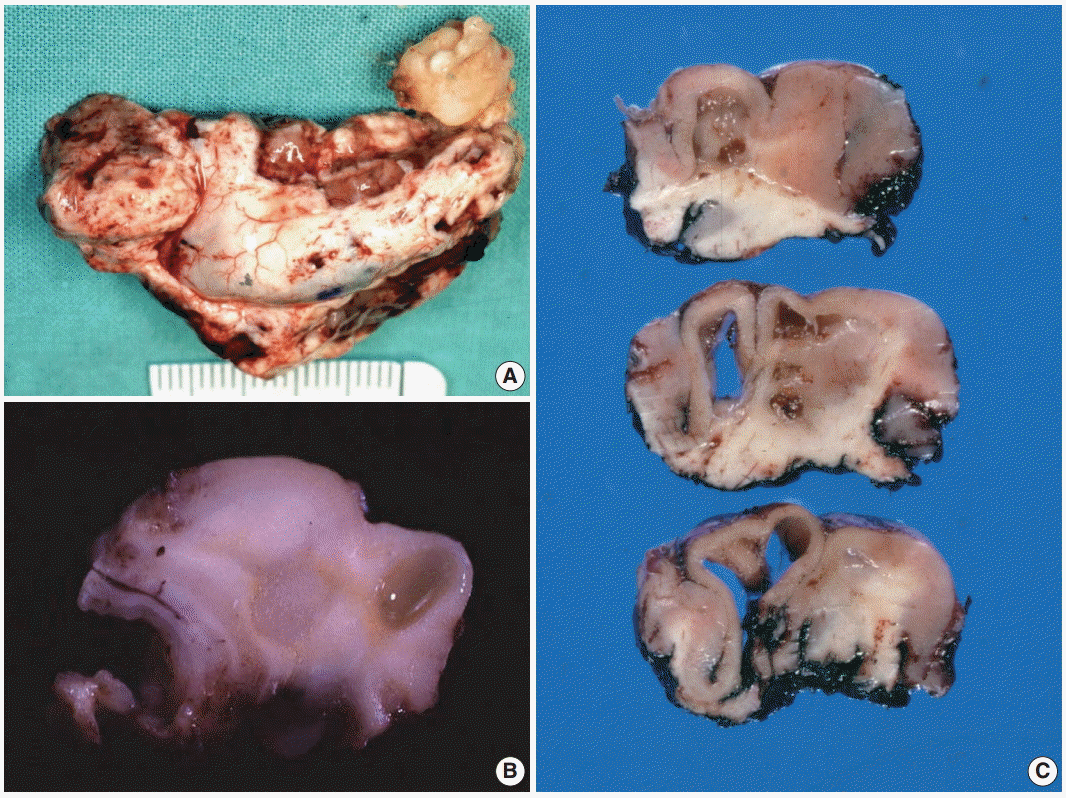 | Fig. 2. Gross findings of dysembryoplastic neuroepithelial tumors (DNTs). (A) A hippocampectomy specimen shows a well circumscribed, gray white mass with two small satellite nodules. (B) On the cut section, a complex type of DNT shows multiple gray-white or gelatinous nodules affecting the cortex and white matter. (C) Nonspecific DNT shows a poorly demarcated, cortical thickening with underlying area of white matter rarefaction and cyst formation.
|
Among three histologic variants of DNTs (Fig. 3), both simple and complex subtypes are characterized by the presence of specific GN elements that consist of small, round monotonous cells, so-called oligodendroglia-like cells (OLCs) and floating neurons in an abundant mucinous matrix. The specific GN elements may form typical nodules (Fig. 3A), but they may also show a diffuse pattern. The OLCs are arranged in a columnar pattern perpendicular to the cortical surface and separated by a mucinous matrix. Depending on the amount of mucinous matrix in the specific GN elements, OLCs may show their arrangement in various patterns including microcystic, alveolar, compact, or targetoid structure (Fig. 3B–D). Floating neurons that are observed within the mucin pool typically lack perineuronal satellitosis or nuclear atypia (Fig. 3E). The simple type of DNT consists only of the specific GN elements, and the complex type additionally has glial nodules and/or FCD of the adjacent cortex. The glial nodules show morphological similarity to low-grade gliomas, including oligodendroglioma, astrocytoma, mixed oligoastrocytoma, pleomorphic xanthoastrocytoma, and pilocytic astrocytoma (Fig. 3F, G). Frank nuclear atypia and multinucleated cells are commonly observed in glial components (Fig. 3H). The internodular areas are frequently abnormal, and may contain oligodendroglial and/or astrocytic components, although in some tumors, they are normal. Nonspecific DNT consists of poorly demarcated, diffuse cortical lesions with blurred normal anatomical landmarks (Fig. 3I). The various glial components seen in this type of DNT are similar to those seen in the glial nodules of the complex forms of DNTs or resemble diffuse components observed in the internodular areas of classical multi-nodular DNTs [4,6,7] (Fig. 3J). Nuclear pleomorphism of glial cells is commonly found in all three types of tumors and predominantly affects the astrocytic component. In one of the largest series studied, rarefaction of the underlying white matter (34%), leptomeningeal involvement (51%), and hemosiderin pigmentation (53%) are frequently found but are less common in simple DNTs. Calcification (25%) is not uncommon. Necrosis, mitosis, and microvascular proliferation are rarely observed [6]. Foci of FCD in the adjacent neocortex are found in roughly two-thirds of DNT cases [11].
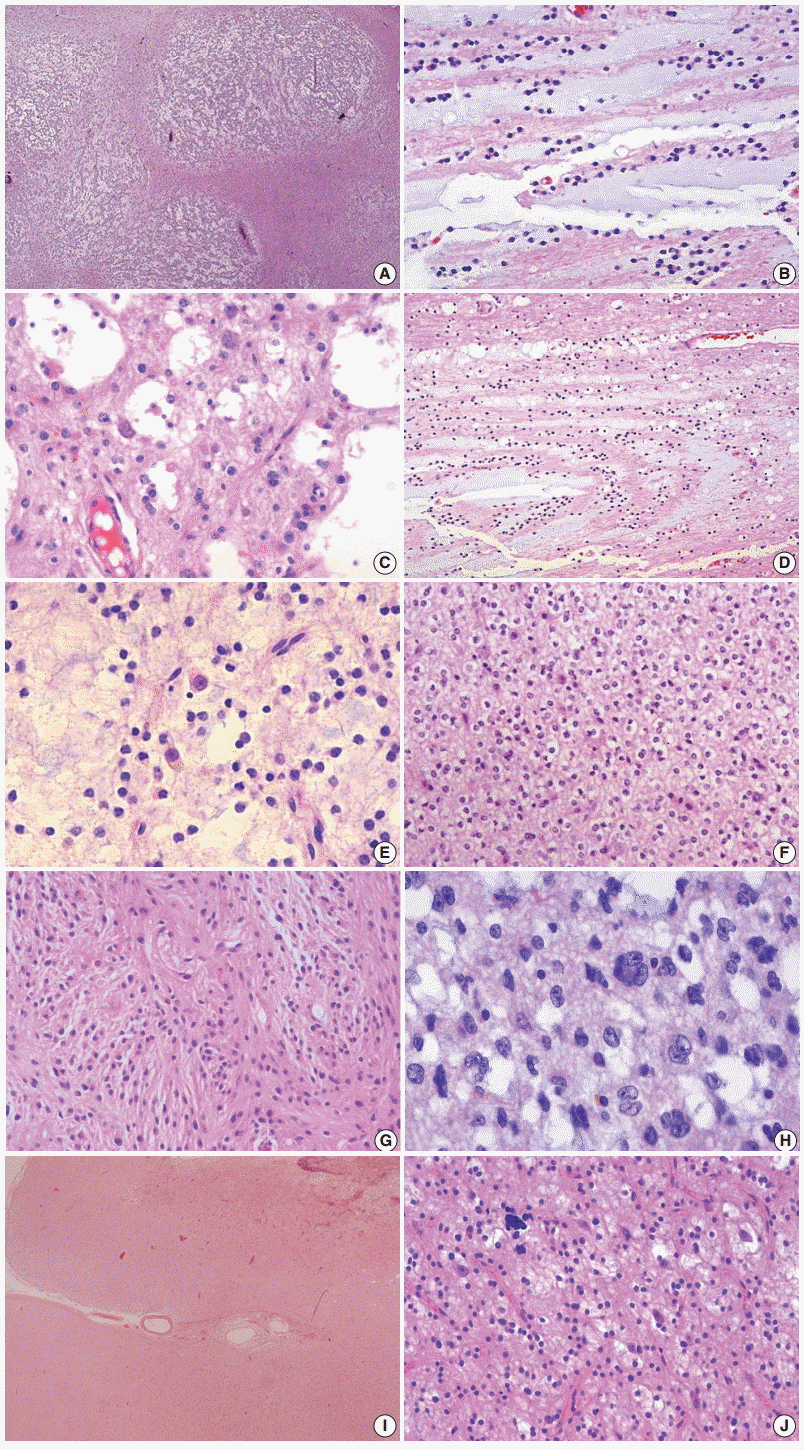 | Fig. 3.Histopathological findings of dysembryoplastic neuroepithelial tumors (DNTs). (A) Multi-nodular appearance typical of complex DNTs. (B) Column arrangement of oligodendroglioma-like tumor cells (OLCs) in the specific glioneuronal element. (C) The characteristic appearance of DNTs with OLCs and mature neurons. (D) Targetoid structure of the specific glioneuronal element. (E) Floating neurons in the mucinous matrix. (F) The histology of glial nodules resembling oligodendroglioma. (G) This glial nodule with predominantly astrocytic differentiation. (H)Nuclear pleomorphism in glial component of DNTs. Histopathological findings of dysembryoplastic neuroepithelial tumors (DNTs). (I) A poorly demarcated cortical lesion of nonspecific DNT. (J) The similar histology of nonspecific DNTs to that observed within the glial nodules of complex DNTs.
|
Go to :
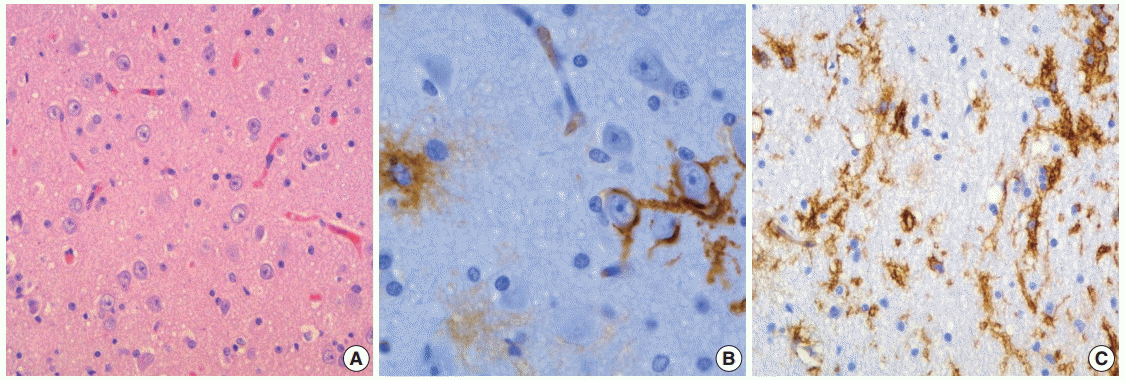
IMMUNOHISOCHEMISTRY
In specific GN elements, floating neurons have expressed neuronal markers including synaptophysin neurofilament, NeuN, neuron specific enolase, MAP2, and class-III beta-tubulin (Fig. 4A, B). The majority of OLCs are strongly positive for S-100 protein and Oligo-2 but generally negative for glial fibrillary acidic protein (GFAP) (Fig. 4C, D). Rarely, OLCs may show immunoexpression of NeuN. Glial nodules contain variable numbers of GFAP-positive astrocytes. In the nonspecific type of DNTs, synaptophysin granular staining is slightly decreased in the lesion compared with that seen in the adjacent normal cortex (Fig. 4E). Other neuronal markers except MAP2 are negative in nonspecific DNTs, but MAP2 is frequently expressed [8]. The expression of CD34 has been reported in 25% to 61% of cases [6-8]. In our study, CD34 expression was more frequently observed in nonspecific types (83.3%) than in simple (10%) and complex types (30.8%). CD34 was positive in the neuronal cell membrane, pericellular stroma, and cytoplasm of OLCs and stellate cells with astroglial morphology (Fig. 4F). CD34 positive cells were focally identified in specific GN elements, whereas most nonspecific tumors and glial nodules of some complex DNTs showed a focal, multifocal, or diffuse pattern of CD34 immunoreactivity (Fig. 4G, H). CD34 was expressed in the peritumoral cortex (Fig. 5A–C), which is more frequent in nonspecific (94.4%) than in classic (26.1%) DNTs. Different expression patterns of nestin and MAP2 in three subtypes of DNTs have also been demonstrated [8,30]. Combined analysis of CD34 and MAP2 is useful in differential diagnosis between nonspecific DNTs and diagnostically challenging mimickers, which will be discussed in the section on differential diagnosis. The Ki-67 labeling index is generally low, below 1% or 2%. The immunohistochemical detection of BRAFV600E has been described in 30% of DNTs, including specific and nonspecific tumors [31]. BRAFV600E immunostaining is diffusely and strongly positive in glial nodules and usually negative in the floating neurons. However, dysplastic neurons show strong BRAFV600E immunoreactivity in complex DNTs associated with FCD.
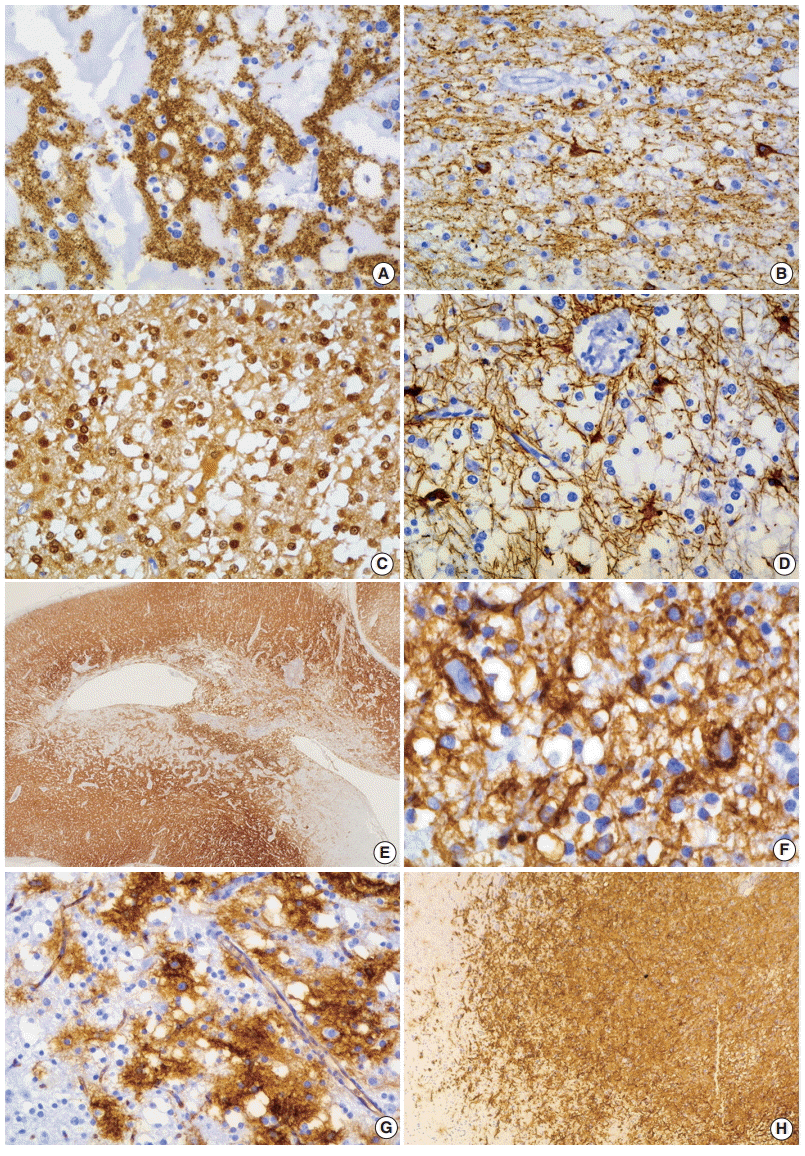 | Fig. 4. Immunohistochemical findings of dysembryoplastic neuroepithelial tumors (DNTs). (A, B) Floating neurons are positive for synaptophysin (A) and phosphorylated neurofilament (B). (C, D) The oligodendroglioma-like cells (OLCs) are diffusely positive for S-100 (C) but negative for glial fibrillary acidic protein (D). (E) Nonspecific DNTs show slightly decreased synaptophysin granular staining compared with that seen in the adjacent normal cortex. (F) In the specific glioneuronal element, CD34 is expressed along the perikarya and in membrane of the floating neurons, pericellular stroma, and cytoplasm of OLCs. (G, H) The specific glioneuronal element shows cluster staining pattern of CD34 (G), while diffuse CD34 immunoreactivity in glial nodules of complex DNTs (H).
|
Go to :
ULTRASTRUCTURAL FINDINGS
Ultrastucturally, OLCs exhibit round to oval invaginated nuclei with evenly dispersed chromatin, marginally condensed heterochromatin, and frequently a single small nucleolus (Fig. 6A). The cytoplasm of OLCs has scanty numbers of organelles, such as rough endoplasmic reticulum, mitochondria, microtubules, and clear vesicles. Unlike oligodendroglial cells, OLCs do not show abundant microtubules, although oligodendroglial differentiation such as pericellular lamination of cell processes has been demonstrated in one case [32]. OLCs may show features of neuronal differentiation [33]. The neuronal features include scant dense core granules (45–60 µm), clear vesicles, synaptic junctions, or neuropil-like cellular processes (Fig. 6B, C). OLCs may show astrocytic features with small numbers of intermediate filaments. Ganglion cells are also observed but never exhibit the dysplastic features or abundant dense core granules of gangliogliomas. Ribosome-lamellae complexes can be found in a few OLCs (Fig. 6D). They are cylindrical and resemble “laboratory tubes with cone-like endings [33].” The presence of these inclusions in OLCs suggests an astrocytic differentiation, because they have been demonstrated in tumors of astrocytic lineage, including glioblastomas.
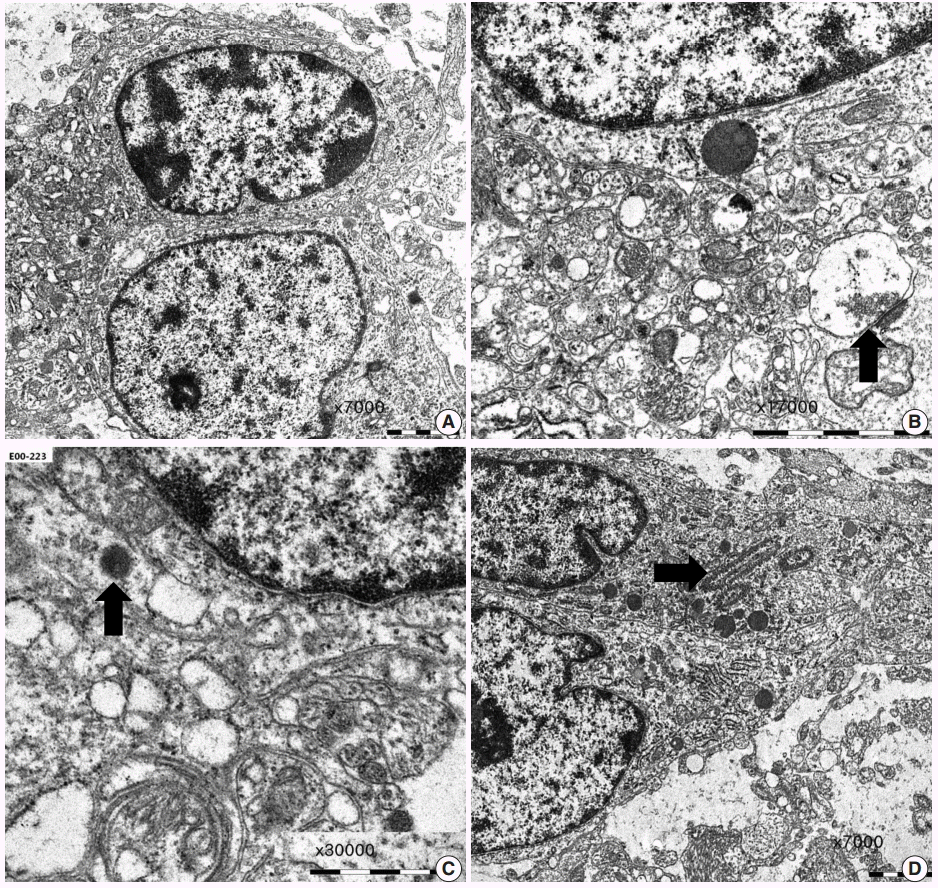 | Fig. 6. Ultrastructural findings of dysembryoplastic neuroepithelial tumors. (A) Oligodendroglioma-like cells (OLCs) show oval nuclei with small indentation, marginal aggregates of heterochromatin, and their cytoplasm with scanty organelles (×7,000). (B, C) Neuropil-like network of cellular process with a synaptic contact (arrow) (B, ×17,000) and scant dense core granules (arrow) (C, ×30,000) indicates neuronal differentiation of OLCs. (D) A few OLCs contain ribosome-lamellae complex inclusions (arrow) (×7,000).
|
Go to :
SQUASH CYTOLOGICAL FEATURES
The cytological features of squash preparations of DNT during intraoperative consultation are fairly characteristic and reliable for correct intraoperative diagnosis, which helps to determine the appropriate neurosurgical procedure. The floating neurons and extracellular mucin that are typical of DNT are more easily demonstrated in cytological preparations than in frozen sections [34] (Fig. 7A, B). OLCs appear as aggregates or are dispersed around abundant arborizing capillaries or in the mucinous or focal fibrillary background. Round to oval naked nuclei of OLCs are larger than non-neopalstic and neoplastic oligodendrocytes. Cytologically, distinguishing features of OLCs from oligodendrogliomas include frequent indentation of the nuclear membrane and multiple, small nucleoli (Fig. 7C). The nuclei of oligodendrogliomas are round with a smooth outline and contain one or two occasional nucleoli (Fig. 7D). The presence of eosinophilic granular bodies in the background favors DNTs rather than oligodendrogliomas.
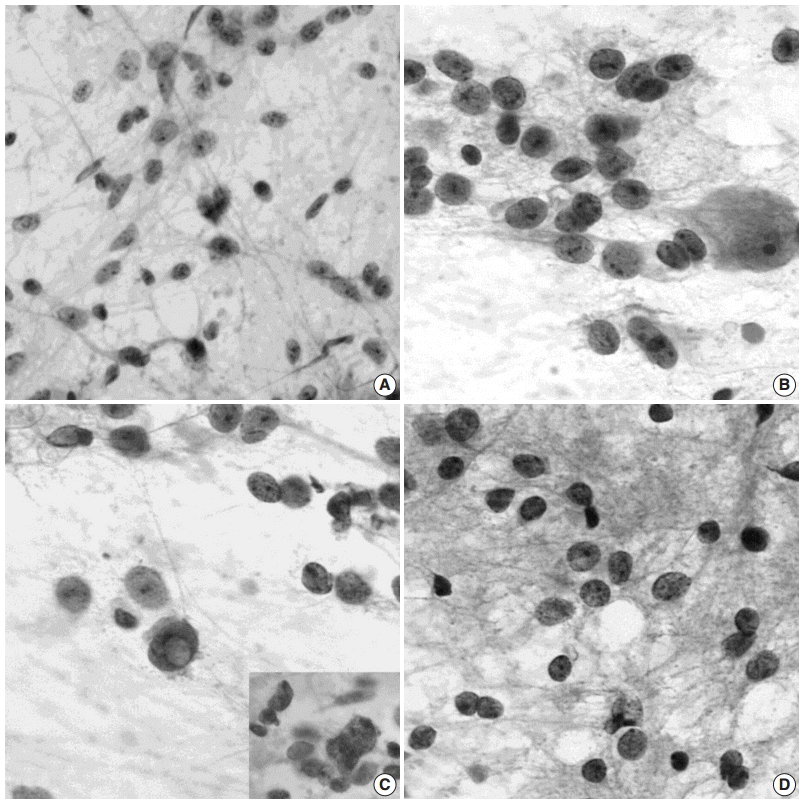 | Fig. 7.Squash cytological findings of dysembryoplastic neuroepithelial tumors. (A) Squash preparation shows round, oval to elongated naked nuclei in the mucinous background. (B) The nuclei of oligodendroglioma-like cells (OLCs) are irregular with small indentations or deep grooves, and fine, granular chromatin and 1–4 small nucleoli. There is a large, normal-looking neuron in the mucinous background. (C) OLCs shows multinuclear giant cell formation (inset) and an intranuclear pseudoinclusion. (D) Squash preparation of oligodendroglioma shows smaller, dark nuclei without nucleoli and larger nuclei with granular chromatin and micronucleoli.
|
Go to :
DIFFERENTIAL DIAGNOSIS
Histologically, the recognition of a unique, specific GN element in brain tumor samples from patients with medically intractable, chronic epilepsy serves as a diagnostic feature for complex or simple forms of DNT. The appreciation of unique histological features may be difficult in fragmented specimens. The nonspecific forms, which lack specific GN elements and multi-nodular architecture, are often not distinguished from epilepsy-associated gliomas. Prominent perineuronal satellitosis and subpial aggregates of tumor cells favor gliomas, although a limited degree of secondary structure formation can be seen in DNTs. Distinguishing DNTs, particularly simple and nonspecific types, from gangliogliomas is often difficult because of overlap in their clinicopathological features, including an association with FCD. Gangliogliomas can be distinguished by the presence of neuronal atypia, perivascular lymphocytic infiltration, and reticulin networks surrounding glial cells. DNTs may rarely show perivascular lymphocytic infiltration and mild neuronal atypia, but they do not show reticulin deposition within tumors.
It is important to differentiate DNTs from gliomas or gangliogliomas that may undergo malignant transformation. Combined immunostains for CD34 and MAP2 can facilitate the differential diagnosis between DNTs and epilepsy-associated tumors that mimic DNTs [8]. Nonspecific DNTs and gangliogliomas share similar immunoreactivity patterns for CD34, but show different immunoreactivity for MAP2 in their glial components. MAP2 immunostaining is faintly positive or negative in the glial component of gangliogliomas, whereas it is frequently seen in DNTs. Most ordinary glial tumors, such as astrocytomas and oligodendrogliomas, are diffusely positive for MAP2 but negative for CD34. The “isomorphic subtype of long-term epilepsy-associated astrocytomas” described by Blumcke et al. [35] shows the absence of both MAP2 and CD34 immunoreactivity. Since BRAFV600E mutations have not been detected in oligodendrogliomas, identification of BRAFV600E mutation by immunohistochemistry or molecular study has diagnostic utility for distinguishing DNT from oligodendroglioma [9].
Go to :
GENETICS
In a study of 13 DNTs, none of the tumors showed 1p19q deletion, IDH1-2, or p53 mutation [36], but a large series of DNTs found IDH1 mutation in three cases, loss of heterozygosity (LOH) 1p/19q in 10 cases, isolated LOH 19q in two cases, LOH 10q (PTEN locus) in three cases, and combined LOH 1p/19q and 10q in one case [6]. Other studies, however, including this one, did not show IDH1 mutation [9,26]. IDH2 mutation or epidermal growth factor receptor amplification has not been identified. Recently, BRAFV600E mutations were identified in 30%–51% of DNTs [9,31]. In our study, the frequency of BRAFV600E in DNTs was related to the location of tumors and was more frequent in extratemporal location (68.2% vs 37.9%). BRAFV600E mutation was more common in classical DNTs (59.4%) than nonspecific DNTs (36.8%). Recently, copy number aberrations (CNAs) have been identified in DNTs, and the most frequent are gains at chromosomes 5 and 7, often concurrent, and at chromosome 6. CNAs in DNTs are not correlated with their histological subtypes, CD34 expression or clinical features [37].
Go to :
HISTOGENESIS
DNTs were initially believed to be a hamartoma in nature [2-4], but recent identification of genetic alterations in DNTs, including classical and nonspecific types, indicate that this tumor is a neoplastic condition. The histogenesis of DNTs remains unknown. The developmental origin from the secondary germinal layer has been proposed based on the mixed cellularity of DNTs, their preponderance in the temporal lobe, and their association with FCD [2]. The origin of DNTs from pluripotent precursor cells is also suggested based on ultrastructural findings of OLCs showing neuronal and glial differentiation. This hypothesis is supported by the immunoexpression of both nestin and MAP2 in OLCs, which has been identified in neural and glial precursor cells during human development. Higher expression levels of nestin, MAP2 and CD34, stem cell markers, in nonspecific rather than simple or complex DNTs suggests an earlier developmental origin of the nonspecific DNTs [8,30].
Because DNTs share CD34 expression, BRAFV600E mutations, and chromosomal copy number profiles with gangliogliomas, and occurrence of composite tumors with ganglioglioma and DNT, both tumors may represent different morphological variants of the same tumor entity [38].
Go to :
 XML Download
XML Download