

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Although uncommon 50 years ago in North America, Barrett esophagus (BE) and BE-associated dysplasia and malignancy have rapidly increased in incidence, such that pathologists in North America encounter screening biopsies for BE routinely, and glandular dysplasia/malignancies much more frequently than squamous lesions. The epidemiology of this trend suggests that issues such as increasing rates of obesity and metabolic syndrome may be part of the cause.1 Although the incidence of BE has been reported to be low in South Asian countries, it appears to be increasing.2 Pathologic evaluation of diagnostic biopsies and therapeutic mucosal resections can be challenging; specific issues are reviewed in this article.
DEFINITION OF BE
The definition of BE varies worldwide, and there is a lack of consensus regarding the importance of identifying intestinal metaplasia, defined histologically by the presence of goblet cells within gastric-type mucosa. The guidelines published by the American College of Gastroenterology defines BE as a change in the distal esophagus, of any length that can be recognized as columnar by endoscopy and showing intestinal metaplasia on biopsy.3 This position is also supported by the American Gastroenterological Association (AGA).4 The histologic demonstration of goblet cells is, however, not required by British and Japanese gastroenterologists.5-7 The British Society of Gastroenterology defines BE as metaplastic columnar mucosa that is clearly visible endoscopically (≥1 cm) above the gastroesophageal junction and is confirmed to be metaplastic with biopsies, but does not require the histologic identification of goblet cells (i.e., gastric-type mucosa with or without intestinal metaplasia).7 The reasoning behind this definition is that the absence of goblet cells is, for the most part, due to sampling and if an adequate number of biopsies are taken, goblet cells can be identified. This is supported by a number of studies.8 In a follow-up study by Gatenby et al.,9 >50% of patients without intestinal metaplasia at index biopsy subsequently showed goblet cells at 5 years follow-up, which increased to 90% at 10 years. Harrison et al.8 demonstrated that the percentage of patients with intestinal metaplasia increased with the number of biopsies taken, and suggested that a minimum of eight samples is required.
BE is important to diagnose because of its preneoplastic nature and risk of progression to adenocarcinoma. The debate regarding the importance of intestinal metaplasia essentially questions the neoplastic risk of columnar mucosa without goblet cells. There are three types of metaplastic columnar mucosa that may replace the normal esophageal squamous epithelium: 1) intestinal type characterized by the goblet cells; 2) cardiac type containing cardiac-type mucous glands; and 3) oxyntocardiac type, which contains a mixture of oxyntic mucosa and cardiac-type glands. Data regarding the neoplastic potential of the intestinal type, as compared with that regarding the non-intestinalized types, are mixed.
Columnar mucosa without goblet cells may in fact show intestinal differentiation. Studies have demonstrated that metaplastic columnar mucosa without goblet cells expresses immunohistochemical markers associated with intestinal differentiation, such as CDX2, villin, and MUC-2, and shows similar molecular and DNA content abnormalities to columnar mucosa with goblet cells.10-12 The view that all metaplastic columnar mucosa has neoplastic potential is supported by a study conducted by German and Japanese pathologists and gastroenterologists. These researchers found that in 70% of small (<2 cm) early adenocarcinomas resected by endoscopic mucosal resection (EMR), the background mucosa was cardiac or oxyntic type rather than intestinal type.13 This study, however, did not report whether intestinal metaplasia was present in other areas of BE without adenocarcinoma, as loss of intestinal differentiation may be seen with neoplastic progression. In addition, clinical follow-up studies involving patients in the UK have shown similar rates of progression to dysplasia and malignancy, regardless of the presence or absence of intestinal metaplasia.9,14 These studies were, however, limited by sampling at index biopsy.
Conversely, other long-term studies support the North American BE definition by showing that when BE is adequately sampled, only patients with goblet cells progress to dysplasia and malignancy. In the study by Chandrasoma et al.,15 patients underwent systematic 4-quadrant, multilevel biopsies taken every 1 to 2 cm throughout the entire visible lesion. Intestinal metaplasia was present in 87.4% of patients, and dysplasia and/or adenocarcinoma was only seen in these patients. Westerhoff et al.16 similarly found dysplasia and cancer only in patients with documented goblet cells and noted that those with short segments without goblet cells continued to not have goblet cells on subsequent biopsies, implying these may actually represent proximal stomach rather than true BE.
Overall, evidence showing the importance of intestinal metaplasia is mixed, and study limitations prevent definitive conclusions. The position statement from the AGA continues to support the presence of intestinal metaplasia, as they believe that this is the only type of esophageal columnar mucosa that clearly predisposes to malignancy.4 The British Society for Gastroenterology stands by the endoscopic diagnosis but recognizes that short tongues without goblet cells are of less clinical significance. This should be taken into consideration in follow-up and surveillance programs; however, they do not specify what that the time interval should be.7
DISTINGUISHING BE FROM INTESTINAL METAPLASIA OF THE CARDIA
Biopsies from the gastroesophageal junction showing intestinal metaplasia may represent BE or intestinal metaplasia of the cardia. Since metaplastic columnar mucosa of the esophagus is histologically similar to the cardia, distinguishing the two histologically can be very difficult, if not impossible in some cases. However, some features can be used to identify the esophageal location. Structures that are native to the esophagus, such as esophageal glands and/or ducts, are most specific. The muscularis mucosa (MM) in BE is often duplicated (further detailed below), which is also a helpful feature that can be appreciated in EMR specimens but not on biopsy. Other histologic features found more frequently in BE include a multilayered epithelium, hybrid glands and squamous islands, or squamous mucosa overlying crypts.17-19 Unfortunately, these features can only be identified in approximately 30% of biopsies; therefore, in most cases, this distinction cannot be made histologically.
Clinically, it is also difficult to distinguish ultrashort BE (<1 cm) from an irregular Z-line, and there is lack of interobserver agreement in diagnosing BE endoscopically until the segment is at least 1 cm in length. In ultrashort BE, the risk of progression is unclear but is thought to be relatively low. Therefore, owing to the inability to distinguish BE from intestinal metaplasia of the cardia histologically in most cases and with lack of evidence on the risk of ultrashort BE and intestinal metaplasia of the cardia, it has been advocated that gastroenterologists should not routinely biopsy the cardia or an irregular Z-line.7,20
HISTOLOGIC DIAGNOSIS OF DYSPLASIA
The categories of dysplasia described in the Vienna classification are used in BE.21 Although defined criteria exist, the recognition and interpretation of these are somewhat subjective, leading to significant intraobserver and interobserver variability in diagnosing all grades of dysplasia. As dysplasia falls along a spectrum, a lack of agreement generally exists between the definitions of low-grade and indefinite for dysplasia, low-grade and high-grade dysplasia, and high-grade dysplasia and intramucosal carcinoma.22 Agreement is often better at the lowest and highest ends of the scale, i.e., negative for dysplasia and high-grade dysplasia. Furthermore, there are significant differences in thresholds between North American and Japanese pathologists, particularly in the diagnosis of adenocarcinoma.23 Owing to the lack of consensus, practice guidelines generally advocate a second review of biopsies with dysplasia by a gastrointestinal pathologist.3,7 It has been shown that the risk of progression is increased when there is consensus on the presence of dysplasia.24,25
Negative for dysplasia
Metaplastic columnar mucosa in the esophagus may show severe reactive and regenerative changes, as it is often inflamed and traumatized by chronic gastroesophageal reflux. The deep glands or crypts may show a degree of crowding and often show nuclear hyperchromasia, enlargement and pleomorphism, particularly the intestinal type, as the nuclei of intestinalized epithelial cells are normally larger and more hyperchromatic than those of gastric-type epithelial cells. Surface maturation is an important and reassuring histologic feature. The surface epithelial cells are nonstratified and have basally located nuclei that show no loss of polarity. Usually, in reactive epithelium, the cytologic atypia is uniform throughout the biopsy specimen without evidence of an abrupt transition.
Low-grade dysplasia
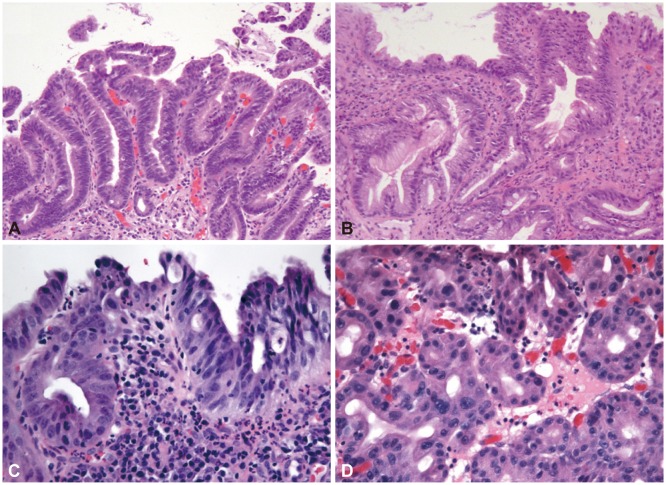
In low-grade dysplasia, nuclei are hyperchromatic, enlarged, and stratified, with this change extending to the surface epithelial cells. Therefore, there is loss of surface maturation. There is only mild or no loss of polarity, no evidence of severe nuclear atypia, and no significant gland crowding (Fig. 1A).
Indefinite for dysplasia
This category is used if the atypia seen cannot be clearly classified as negative or dysplastic, which is usually of low-grade type. As stated previously, the epithelium in BE may show severe reactive/regenerative changes with cytologic features that may overlap with low-grade dysplasia. Usually, the cause for diagnostic difficulty is severe inflammation (Fig. 1B); however, this category may also be used if there are other limiting factors such as crush artifact, poor orientation, or small biopsies preventing the unequivocal diagnosis of dysplasia.
Crypt dysplasia
Crypt dysplasia is diagnosed when dysplasia is present within the basal crypts but surface maturation is seen. As stem cells reside within the crypts, it is plausible that dysplasia would commence there, and this finding may be a risk factor for progression. A study by Lomo et al.26 showed that dysplastic crypts have increased p53 positivity, elevated MIB-1 proliferation rate, and aneuploidy for 17p (TP53), compared with nondysplastic epithelium. A study on the interobserver agreement of this diagnosis found a moderate level of agreement both before and after a consensus conference.27 Although crypt dysplasia may represent a distinct diagnostic entity, it is rarely seen on its own without dysplasia in other surrounding biopsies, and the clinical implication of a diagnosis of crypt dysplasia alone is not currently known. At our institution, we examine deeper sections if crypt dysplasia is seen, and if an alternate diagnosis remains unclear, indefinite for dysplasia is diagnosed, which leads to follow-up biopsies.
High-grade dysplasia
In high-grade dysplasia, there is increased nuclear enlargement with loss of polarity, marked nuclear pleomorphism, nuclear stratification to the luminal surface, and surface mitotic activity (Fig. 1C). Architectural changes are also present with gland crowding and branching, but not to the degree seen in features suspicious for carcinoma (discussed below).
Foveolar dysplasia
A newly recognized type of dysplasia known as foveolar or nonadenomatous dysplasia is typically high grade but may also be low grade. Unlike the intestinal type that has elongated, pencil-like nuclei, in the foveolar type of dysplasia, nuclei are round and basally located with abundant eosinophilic or mucinous cytoplasm. Grading is less well defined but the distinction between low- and high-grade dysplasia rests mainly on increased nuclear size and architectural crowding in high-grade dysplasia (Fig. 1D).28 Nuclear pleomorphism is minimal, even in high-grade dysplasia, although nucleoli may be prominent. This monotony may impart a more bland appearance, which causes difficulty in distinguishing this from reactive mucosa. An important feature in distinguishing reactive change from foveolar dysplasia is the full thickness mucosal involvement in foveolar dysplasia as opposed to surface involvement in reactive change secondary to reflux.29
Intramucosal adenocarcinoma and features suspicious for intramucosal carcinoma
The diagnosis of intramucosal adenocarcinoma on biopsy can be challenging, which has led to the recognition of a number of histologic features deemed suspicious for intramucosal carcinoma (IMCa). The predictive value of these features was examined in a series of preoperative biopsies paired with subsequent esophagectomy specimens.30,31 Histologic features considered to be suspicious for carcinoma included dilated dysplastic glands with necrotic debris, solid or cribriform growth, ulcerated high-grade dysplasia, neutrophils within high-grade dysplasia, and high-grade dysplastic glands invading into the overlying squamous mucosa.29,30 The risk of adenocarcinoma increased significantly if two or more of these features were seen,30 and the presence of three or more dilated glands with necrotic debris was shown to be an independent predictor of adenocarcinoma on resection.31
Patterns of invasion characteristic of intramucosal adenocarcinoma include single-cell infiltration, angulated/abortive glands, sheet-like growth, never-ending/anastomosing gland pattern, and highly complex cribriform arrangement of glands.
As stated previously, there is poor interobserver agreement between North American and Japanese pathologists in the diagnosis of carcinoma. Several of the features described above as high-grade dysplasia or as being suspicious for carcinoma would be diagnostic for carcinoma for several Japanese and some German pathologists.23 Takubo et al.23 state that histologic photographs labeled as high-grade dysplasia and some as low-grade dysplasia in textbooks of surgical pathology in North America and Europe would be diagnosed as clear-cut cases of adenocarcinoma with or without stromal invasion by Japanese pathologists.
Submucosal invasion
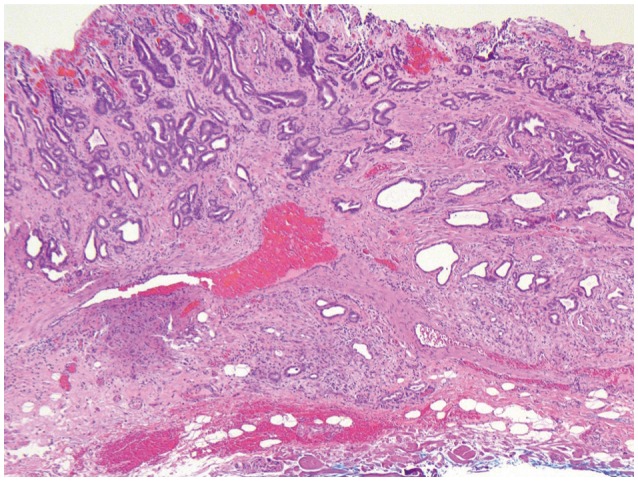
Submucosal invasion is diagnosed when tumor invades through the MM (i.e., the true MM and not the duplicated muscle, as discussed below). Unlike intramucosal carcinoma, submucosal invasion is often accompanied by a desmoplastic reaction. In addition to desmoplasia, tumor adjacent to a large vessel (although not entirely specific) may also signal submucosal invasion (Fig. 2). In equivocal cases, immunohistochemistry for desmin, highlighting the muscle layers, can be useful in identifying the deep limits of the MM (Fig. 3). Distinguishing intramucosal from submucosal invasion is clinically important as the risk of lymph node (LN) metastasis greatly increases.
BIOMARKERS IN BE-ASSOCIATED DYSPLASIA AND MALIGNANCY
In the context of pathology specimens, biomarkers are most commonly detectable proteins, but occasionally may be DNA markers (i.e., aneuploidy) or RNA sequences such as mi-RNA. In BE, there has been considerable investigation of biomarkers as aids in diagnosis, to identify cases at risk of progression and as prognostic and predictive factors in malignancy.32
Considering the difficulties in making confident diagnoses of BE-associated dysplasia in the background of inflammation, the identification of reliable biomarkers would be of considerable use for routine diagnostic purposes. Difficulties with assessing biomarkers in these circumstances are predominantly in study design; biomarker utility is routinely based on the diagnosis of H&E-stained sections, which is inherently unreliable.33
p53 is a transcription factor that acts as a tumor suppressor gene; when a cell is sufficiently damaged, p53 initiates apoptosis. Inactivating mutations of the gene are found in many malignancies and can be detected by immunohistochemistry, which shows either complete loss (absent protein/epitope) or, more commonly, increased expression (due to mutations creating a protein resistant to degradation). In our experience, performing immunohistochemistry for p53 is only rarely useful in the diagnosis of dysplasia, as it is most commonly found to be abnormal in high-grade dysplasia, which is less commonly a diagnostic dilemma than making the diagnosis of low-grade dysplasia vs. reactive changes. However, studies have suggested that cases of low-grade dysplasia that show p53 abnormalities may be at higher risk for progression to malignancy.34,35 p53 may therefore be of greater use as a marker of disease progression. Care must be taken in interpretation of the stain; weak to moderate staining can be seen in scattered nuclei in reactive changes. True abnormalities are present when there is strong, diffuse staining or diffusely absent staining of the nuclei (Fig. 4A).36
Assessment of mitotic activity by staining for Ki-67 can be useful in the diagnosis of dysplasia-the normal active mitotic zone in BE mucosa can be at the base of the crypts (as it is in small intestinal mucosa) or at the neck zone (as it is in the gastric mucosa), depending on the type of mucosa present and the degree of intestinal metaplasia. However, identification of mitoses along the surface of the epithelium is abnormal, except in the case of regenerative epithelium. Although these mitoses can sometimes be easily seen on H&E-stained slides, immunohistochemistry for Ki-67 can readily demonstrate mitotically active cells along the surface, which may allow pathologists to distinguish reactive from dysplastic changes (Fig. 4B).37
Expression of racemase (α-methylacyl coenzyme A racemase, AMACR) has been proposed as an aid to dysplasia diagnosis, as it has been noted to increase progressively as mucosa progresses from BE to dysplasia to malignancy, with considerable variability in the degree of staining between studies.38-40 The increasing proportions of cases staining as one looks at negative, indefinite, low-grade, and high-grade dysplasia indicate that this may not be of significant utility in individual cases-we have not found this to be of use in routine practice. AMACR may be of more utility as a method of predicting which cases may progress, either from indefinite for dysplasia to definite dysplasia or to high-grade dysplasia or to malignancy.41,42 However, interpretation of the stain may itself have interobserver variability, as the study by Kastelein et al.41 proposed a 3-point scale of no staining, mild staining, and strong staining, which could be subject to considerable variability of interpretation.
As noted above, both AMACR and p53 may be biomarkers that can aid pathologists in detecting cases of dysplasia with a higher risk of progression. Various other biomarkers have been suggested (e.g., p16, loss of heterozygosity, aneuploidy, tetraploidy, and epidermal growth factor receptor; reviewed in Varghese et al.32); however, none of these have been validated to the point that they are in routine clinical use. Similarly, factors predictive of response to therapy have been proposed (Ki-67, loss of p16)43,44 but are not yet accepted in routine practice. Prognostic markers for adenocarcinoma are similarly not in common use; however, recent sequencing studies have identified a multigene signature that may be useful.45
In BE-associated adenocarcinomas, human epidermal growth factor receptor 2 (HER2) overexpression represents the major therapeutic biomarker. In the original trastuzumab for gastric cancer trial, gastroesophageal junction adenocarcinomas demonstrated a higher proportion of HER2 amplification than gastric carcinomas; this expression indicated tumors eligible for trastuzumab therapy, and resulted in extended survival.46 Clinical studies on HER2 expression in more proximal esophageal adenocarcinomas are ongoing.
Overall, BE-associated dysplasia and malignancy clearly have considerable heterogeneity on the genetic level, which underlies the problem of identifying biomarkers that apply to an overall population. However, a number of population-based studies are ongoing that will hopefully identify biomarkers with clinical and pathologic utility for these entities.
EVALUATION OF EMRs
EMRs for BE-associated dysplasia and malignancy are processed in the same manner as EMRs performed for squamous dysplasia. Briefly, the specimen should be pinned on a cork or styrofoam board to prevent curling, and fixed in an appropriate volume of 10% formalin. In the pathology laboratory, the deep margin should be stained with ink or silver nitrate before serially sectioning the specimen and embedding. In our laboratory, we routinely cut four sections from each block of tissue: two for H&E staining; one for hematoxylin, phloxine, and saffron staining, to aid in evaluating the muscle layers; and one for alcian blue stain, to aid in identifying goblet cells.
An issue that arises in EMRs taken for BE-associated dysplasias and malignancies that is not identified in those done for squamous lesions is that of the duplicated MM (dMM) (Fig. 3). The observation that the MM is frequently present as a dual-layer underlying BE mucosa was first identified in the 1980s in esophagectomy specimens47-49 and has been referred to as a musculo-fibrous anomaly. This seems to be exclusive to BE in the esophagus; it was not identified in 352 cases without BE in the esophagus48 and has not been reported to occur as underlying intestinal metaplasia in the stomach. In esophagectomy specimens, this is a frequent finding, with 46% to 100% of specimens demonstrating a dMM,47,49-52 which averages to approximately 80% of these specimens containing dMM. Few papers have evaluated the presence of dMM in EMR specimens; this suggests that EMRs overall show a lesser proportion of tissue with a dMM, with 46% and 66% containing dMM.53,54 This most likely relates to the focality of this change. In esophagectomy specimens, reports indicate that, whereas duplication is noted in most specimens, it is not present along the entire length of the BE segment: in various reports, dMM is found along 5% to 90% of this length.49,51 In EMR specimens, it is more difficult to evaluate the proportion of the overall BE segment with dMM; however, one study noted that 46% of specimens had focal or extensive dMM (10% to 100%), whereas 54% had minimal (<10%) or absent dMM.53 A separate study on EMRs identified extensive duplication in 38% of the specimens, moderate in 33%, and minimal in 29%.54 On reviewing a series of cases from St. Michael's Hospital, in 106 EMR specimens from 30 patients, at least focal dMM could be identified in 101 cases (95%). To add to the difficulty, some cases can show triplication of the MM, and some do not show true duplication but extensive thickening of a single layer of muscle.51 Clearly, dMM is frequently present; however, it may be patchy enough that it cannot reliably be found in all slides from EMR specimens. The cause of this stromal alteration has not been identified; whether this is a metaplastic phenomenon occurring along with the metaplasia within the mucosa, or is simply due to splaying and hypertrophy of the MM secondary to inflammation and ulceration has not been evaluated. The significance of the presence of dMM has to do mainly with the determination of the presence versus absence of invasion as well as the depth of invasion. With respect to presence/absence of invasion, the early term musculo-fibrous anomaly was used as a descriptive term, as this change frequently has associated fibrosis along with the duplication and hypertrophy of the MM, with frequent ingrowth of smooth muscle fibers into the lamina propria of the mucosa, in a manner very similar to mucosal prolapse changes seen elsewhere in the luminal gastrointestinal tract.47 These fibers within the lamina propria often surround small glandular elements, which can respond by distortion and cystic dilatation; this can mimic early invasion by a well-differentiated adenocarcinoma. This is most particularly a problem in biopsy specimens but can also be a problem in EMR tissues. This also raises difficulties in evaluating the depth of invasion in adenocarcinomas. The presence of dMM receives little attention in residency training or in textbooks, and many pathologists are unaware of its existence. This can lead to overstaging of adenocarcinomas that extend into the space between the layers of MM; pathologists may interpret this as submucosal invasion. It is therefore necessary to be aware of this change and to be able to differentiate between the tissue between the two layers of muscle and the true submucosal tissue. The space between the two layers of the MM has the characteristics of lamina propria, with delicate, thin-walled blood vessels.55 In contrast, the submucosa has sturdy, thick-walled muscular arteries in loose connective tissue-these are often present in EMR specimens and are the most reliable indicator of the presence of submucosa. The submucosa also contains submucosal glands: these are very rarely identified in biopsy specimens but are more often present in good quality EMR specimens and in sections from esophagectomies. In addition, invasive carcinomas that are limited to the mucosa (including both layers of the MM) are generally not associated with desmoplasia; although there are occasional reports of desmoplasia associated with IMCa,51,56 well-developed desmoplasia in invasive carcinoma should raise the suspicion of a submucosally invasive carcinoma (Fig. 2). This has significant implications for treatment, as the depth of invasion is significantly linked to prognosis. In squamous carcinomas, the depth of invasion is generally separated into tumor limited to the epithelium (M1), tumor invading the lamina propria (M2), tumor invading MM (M3), and then submucosal invasion divided by depth of invasion into SM1 to SM3.57 This correlates well with the increasing risk of LN metastases, with M1 tumors having 0% risk, M2/3 tumors having approximately 4% risk, and submucosally invasive cancers having risks of LN metastases increasing from 24% to 48% as the depth of invasion increases. In BE-associated adenocarcinomas, evaluation of the depth of invasion is complicated by the presence of dMM. In our practice, we use the staging system proposed by Vieth and Stolte.57 In this system, M1 indicates lamina propria invasion, M2 invasion of the superficial/duplicated layer of the MM, M3 invasion into the space between the layers of MM, and M4 invasion of the deep/true MM. Then, submucosal invasion is staged in the same manner as invasive squamous cell carcinoma. This more extensive separation of IMCa by the layers does not predict any differences in the rate of LN metastases,51,54,56,57 most probably because the presence of lymphatic spaces is similar throughout the layers. However, we find that this mode of separating the depth of invasion most clearly expresses the depth of invasion to our gastroenterology colleagues. It also reminds us to consider the presence of a dMM to prevent overstaging of adenocarcinomas as submucosal invasion (T1b) when it remains restricted to the mucosa (T1a), a significant prognostic factor.
BEYOND EMR TREATMENT: WHEN IS AN ESOPHAGECTOMY NECESSARY?
In T1a adenocarcinomas that are well-differentiated or moderately differentiated and show no evidence of lymphovascular space invasion, it is generally assumed that the risk of LN metastases is sufficiently low (<5%) and that the risk of esophagectomy is greater than the gain from surgery. EMR therapy and/or other mucosal ablation techniques such as radiofrequency ablation are often curative in these patients.58 Long-term follow-up for these patients is necessary, as they may have residual/recurrent mucosal dysplasia or malignancy. Even early submucosal invasion (SM1) seems to have a good outcome in most patients when treated with curative EMR.59 However, these good outcomes are limited to low-risk malignancies: no deeper than SM1 invasion and well-differentiated to moderately differentiated tumors, which are often small and show no evidence of lymphovascular space invasion. It becomes significantly more difficult to determine the proper course of treatment in cases with additional poor prognostic features, such as lymphovascular space invasion, poor differentiation, tumor budding, or micropapillary architecture. The prognostic significance of micropapillary architecture has not been described in tumors of the gastroesophageal junction but has been associated with increased risk of LN metastasis and poor prognosis in gastric tumors.60-62 Tumor budding has also been associated with poor prognosis in both squamous and adenocarcinoma of the esophagus; however, its independent prognostic significance in early tumors is unclear.63 While at this time, there are limited data on the significance of these poor prognostic factors in early tumors, these features should be identified by the pathologist as they may suggest a more aggressive course and a surgical consultation may be warranted.
CONCLUSIONS
The incidence of adenocarcinomas of the distal esophagus and gastroesophageal junction appears to be increasing, not only in North America but now in South-East Asia as well. Many of these arise in the background of BE; in North America the diagnosis of this most often rests on the histologic observation of goblet cells within columnar type mucosa. As BE progresses toward dysplasia and malignancy, there is considerable interobserver variability between pathologists in diagnosis. Currently available biomarkers are of little utility in aiding diagnosis. An important factor to consider when evaluating the depth of invasion of mucosal resections of these adenocarcinomas is the presence of the duplicated muscularis mucosae. Definite submucosal invasion or the presence of adverse histologic factors such as lymphovascular space invasion should lead to the consideration of esophagectomy.


 XML Download
XML Download