

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cowden's disease (CD), first discovered and described as a disease by Lloyd and Dennis1 in 1963, is a rare hereditary genodermatosis disorder. It is characterized by germ line mutations in the phosphatase and tensin homolog (PTEN) on chromosome 10q22-q23 tumor suppressor gene.1,2
CD is associated with multiple hamartomas of ectodermal, mesodermal, and endodermal origins as well as increased risk of developing breast, thyroid, and endometrial cancers.3 The development of the mucocutaneous lesions usually precedes the malignancies in most of the cases, which is why early diagnosis of the lesions is critical. Once a definite diagnosis is made, appropriate primary screening focusing on the cancer risks should be carried out according to the CD guidelines, as described in the National Comprehensive Cancer Network (NCCN) website. Proper prescriptions to the family members should also follow.4
Here, we report our novel findings of the gastrointestinal (GI) endoscopies in a family with CD, including a daughter with a history of surgery for thyroid cancer due to the PTEN mutations inherited from her paternal family.
CASE REPORT
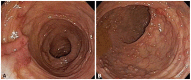
A 52-year-old male patient without any relevant medical history visited the hospital with a symptom of recent lower abdominal pain. The patient had been smoking for 35 years and had a past history of heavy alcohol consumption. He received a lipoma surgery for a palpable mass pressure in the anterior neck portion 3 years ago. For the first time in his life, the patient received colonoscopy, which showed multiple polyps of different size and shape in the terminal ileum, the colon and the rectum (Fig. 1), especially hundreds in the rectum. Several biopsies were taken throughout the entire large bowel including the terminal ileum, and they were histopathologically confirmed as inflammatory and hyperplastic polyps. Physical examination revealed that the head circumference of the patient was 630 mm (for reference, the average and 99 percentile occipitofrontal circumferences of Korean males between 50 and 59 years old are 568 and 601 mm, respectively), suggesting macrocephaly. Also black pigmented cutaneous papules could be seen on the lips and multiple soft humps were found in the posterior neck, arm, and hip. The neck computed tomography (CT) revealed dense fat masses in the posterior neck as well as in both the submandibular and supraclavicular areas (Fig. 2). The upper GI endoscopy revealed multiple mucosal papillomas at the gingiva, oral cavity, soft palate, pharynx, and epiglottic areas (Fig. 3). There were also elevated flat lesions of whitish color in the esophagus, and biopsies showed they were squamous acanthosis. The multiple round gastric and duodenal polyps of various sizes were turned out to be hyperplasia (Fig. 4).
Considering all symptoms and the results of the examinations, CD was the most likely clinical diagnosis. Imaging studies of the brain, thyroid, chest, and abdomen as well as PTEN gene mutation test were performed. At thyroid ultrasonography (USG), an oval shaped isoechoic nodule of 1.2 cm in diameter was found in the isthmus. The fine needle aspiration (FNA) was consistent with nodular hyperplasia. The abdominal CT scan presented hemangiomas approximately 2 and 2.7 cm in size in both lobes of the liver, and a gallbladder stone of 7 mm in size. The brain magnetic resonance imaging (MRI) and the chest CT did not show any mass or abnormal finding. The laboratory test results were also within the normal ranges.
The examination of the genomic DNA showed a known mutation, c741dupA, in exon 7, which is registered in the Human Gene Mutation Database. It was predicted to lead the frameshift mutation that results in the formation of the premature stop codon (p. Pro248ThrfsX5) for the PTEN protein translocation.
The study was extended to the patient's daughters in order to carry out the adequate screening. The eldest daughter, who was 28-year-old at the moment, received a total thyroidectomy with left modified radical neck dissection and high dose 131I therapy (150 mCi) about a year ago. At that time, thyroid USG showed that there were three thyroid nodules and abnormally enlarged multiple lymph nodes in the left upper and mid jugulodigastric chain. FNA revealed papillary carcinoma with lymph node metastases. Biopsy of the surgical specimen showed multifocal intrathyroidal papillary carcinoma and Hashimoto's thyroiditis with the right perithyroidal and the left level IV and V regions lymph nodes metastases (Fig. 5). Breast USG showed multiple cysts of well-defined smooth borders which were less than 1 cm in diameter and a septated cystic mass which was 2.8 cm in diameter. Core needle biopsy of the 2.8 cm sized cystic mass at the outer area of the lower right quadrant revealed intraductal papilloma. She also had the head circumference of 610 mm in diameter, indicating macrocephaly (The average and 99 percentile occipitofrontal circumferences of Korean females aged between 25 and 29 years old are 552 and 583 mm, respectively). The black pigmented lip, the multiple papillomas of the gum and the soft tissue mass in the size of 9×6×5 cm on the right side of the posterior axillary area, were also found, and the excisional biopsy of the soft tissue mass revealed lipoma. GI tract examination, which was the first time in her life, showed multiple acanthosis lesions in the esophagus, numerous polyps in the second and third regions of duodenum, one fundic gland polyp, and the presence of a lot of inflammatory and hyperplastic polyps in the terminal ileum and the rectum.
In case of the second daughter who was 25-year-old at the moment, there were no suspicious results on the physical examination, GI tract endoscopy, USG of breast and thyroid, and PTEN mutation test was negative.
DISCUSSION
CD, an autosomal dominant disorder provoked by germline mutations of the PTEN tumor suppression gene on 10q23.3, is a rare disease with estimated prevalence of 1 in 250,000 to 200,000, manifested by hamartomatous growths affecting derivatives of all three embryonic origins responsible for a group of phenotypically diverse conditions.5,6
CD is associated with benign findings of variable mucocutaneous lesions which include trichilemmomas, papillomatosis, papules, pigmentations, macrocephaly, lipomas, neuromas, hemangiomas, fibromas and GI hamartomas, fibrocystic breast disease, uterine fibroids, thyroid lesions (e.g., adenoma, nodules, and goiter), and malignancies (e.g., breast, thyroid, and endometrial cancers).4
The diagnosis is based on pathognomonic signs or major and minor criteria of clinical features alone; chromosomal analysis for defective PTEN tumor suppression gene also serves as a useful source of diagnosis. CD can be diagnosed either when any of the pathognomonic criteria are fulfilled or when the proper combinations of the major and minor criteria are satisfied.7 The consensus criteria are described on the website of the NCCN Genetic/Familial High-Risk Assessment Panel (www.nccn.org).
When an at-risk individual, who have any relative with a clinical diagnosis of CD, meet any single major criterion or two minor criteria, CD can be diagnosed without the genetic testing.4 The proband of our case met three major criteria and two minor criteria in addition to the PTEN mutation. Just having nonmedullary thyroid cancer was enough to diagnose the eldest daughter as a CD patient, but she also satisfied four major and two minor criteria.
Even though the clinical symptoms and test results were similar between the father and the daughter, the inspective clinical phenotyping of this family showed that varied features representing age-related penetrance could be seen in different ages. This is a well-known phenomenon in other autosomal dominant tumor suppressor disorders such as neurofibromatosis type 1 as well.8 If the at-risk individual had been investigated during infancy or childhood, many features of CD would not be obvious at that time, and it may take years for the characteristic features to come out, which is why appropriate screening and follow-up observations are required for early detection of the disease.
Macrocephaly can be a clue to early diagnosis of CD. Previous reports of CD have suggested that only a fraction of the patients have the macrocephaly. Unfortunately, however, a lot of these studies did not report the head circumferences of their patients. Lachlan et al.8 reported that macrocephaly was observed in all PTEN carriers including babies, suggesting that PTEN is a negative cellular growth regulator expressed in the brain. The authors also reported that it is important to note macrocephaly antenatally, and that the occipitofrontal circumference is a helpful diagnostic pointer, particularly when deciding which patient is to be tested for PTEN mutation, considering the high cost of the procedure.8
Another reason behind the need of early detection of the disease and observation of the symptoms is the risk of developing malignancy. The most critical forms of CD are presented as breast cancer (life time risk of 25% to 50%), thyroid cancer (life time risk of 5% to 10%), and uterus cancer. However, patients with PTEN mutations are not associated with colorectal or other GI cancers.9 GI observation is not one of the recommended procedures for CD since the relationship between CD and malignant GI tumor is not clearly established yet; however, some reports describing GI cancer in CD have been published.10 Most individuals with CD will have GI polyps with varied histopathological features, which are originally estimated to occur in 60% of the affected individuals, most prevalently in the stomach, and then the colon, esophagus, and duodenum in the same order. Regular colonoscopy is recommended at least at the age of 40 after the baseline screening at some hospitals;9,11 however, earlier and more frequent colonoscopy might be necessary in some cases like the eldest daughter in this case, because she was found to have multiple polyps at the age of 28. Medical attendants of CD patients must be well informed about the manifestations of the GI tract.
The practice guidelines emphasize the importance of examinations: breast self-exam training at the age of 18, clinical breast examination in addition to annual mammography and breast MRI starting at the age of between 30 and 50, or 5 to 10 years younger than the earliest diagnosis in the family, yearly thyroid USG starting at the age of 18, endometrial biopsies beginning between the ages of 35 and 40, or 5 years younger than the earliest diagnosis of endometrial cancer in the family.4,12
Though rare, miscellaneous tumors including meningioma, liposarcoma, non-Hodgkin's lymphoma, and Merkel cell carcinoma were mentioned in the literature in the context of CD.13
PTEN, also known as PTEN on chromosome 10, interacts with focal adhesion kinase, which results in the inhibition of cell migration and spread. It regulates or inhibits extracellular matrix-dependent phosphatidyl-inositol-3-kinase/Akt signaling pathway, which controls the level of phosphoinositol triphosphate and can induce cell cycle arrest in the GI phase and apoptosis as well. The mutation of PTEN tumor suppressor gene that is present on one allele in every cell in a body cannot inhibit the endothelial tube formation induced by the vascular endothelial growth factor (VEGF) which results in the subsequent VEGF expression. The somatic PTEN mutations do not have an antiangiogenic effect, acting as a regulator of the cellular growth activated by tyrosine kinase.14,15
Therefore, the somatic PTEN mutations induce diverse malignancies and are responsible for PTEN hamartoma tumor syndrome (PHTS), which includes CD, Bannayan Riley Ruvalcaba syndrome (BRRS), and Proteus syndrome (PS).14 The PTEN gene mutations are found in up to 80% of the individuals with CD, 60% of BRRS patients, 50% of Proteus like syndrome patients, and 20% of PS patients. The PTEN testing for the specific familial mutation is actively available and can be used to confirm the diagnosis of PHTS.7 The genetic test of PTEN should be advised to all at-risk relatives when the deleterious familial PTEN mutation is known to exist. When the existence of familial mutation is not known but the diagnostic criteria is satisfied, the genetic test should be considered for the affected family members with the highest likelihood of PTEN mutation. Extensive work-up is essential in high risk individuals considering the heterogenous nature and the varied onset age of CD.9




 XML Download
XML Download