

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hypereosinophilia is associated with many allergic, infectious, neoplastic, and idiopathic diseases, and the disease course can range from self-limited to life-threatening disease. Because hypereosinophilic syndrome (HES) can involve multiple organs, it can be fatal. Although the diagnostic criteria for HES should include hypereosinophilic period for at least 6 months, early initiation of therapy may be recommended in HES patients with symptoms.1,2 We report a case of HES with gastrointestinal (GI) involvement treated with early initiation of systemic corticosteroid.
CASE REPORT
A 56-year-old man visited the hospital with complaints of more than 30 bloody diarrheas per day, combined with lower abdominal pain and febrile sensation for 5 days. GI symptom of alternating constipation and diarrhea had persisted for past 3 months. He also had respiratory symptom of chronic cough. He was taking no medications, including herbs and drugs, and had no history of food or drug allergies.
On admission, his blood pressure was 120/80 mm Hg, pulse rate 85/min, respiratory rate 20/min, and body temperature 37.0℃. Physical examinations showed moderate tenderness of the lower abdomen. White blood cell count was 21,040/mm3 with markedly increased eosinophils (11,151/mm3, 53%). Other serum chemistries were normal. Negative test results were obtained for viral markers (hepatitis A, B, C, adenovirus, and human immunodeficiency virus), serum antibodies to parasites, and autoimmune antibodies (anti-nuclear, anti-dsDNA, and anti-neutrophilic cytoplasmic antibodies), tumor markers (alpha-fetoprotein, carbohydrate antigen 19-9, and carcinoembryonic antigen), and rheumatoid factor. No pathogens were detected in a microbiological blood culture, and stool examinations for parasites and protozoa were negative. Simple chest and abdomen X-ray studies were normal.
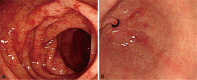
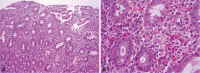
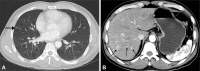
The colonoscopy revealed multifocally erythematous and edematous mucosal changes with disappearance of normal vascular markings and easy-touch bleeding throughout the entire colon, including the rectum (Fig. 1A). There was intense infiltration of the mucosa by eosinophils in the specimens obtained from colonic mucosa (Fig. 2). Although esophagogastroduodenoscopy showed only erythematous change in the antrum of the stomach (Fig. 1B), microscopic findings of specimens from random biopsies of the stomach and duodenal mucosa were similar to those of the colon. Chest computed tomography (CT) for evaluation of the chronic cough showed multiple nodular opacities on the both lung, suggesting eosinophilic pneumonitis (Fig. 3A). Abdominal CT suggested a diagnosis of multiple eosinophilic liver abscesses, which were multifocal, patchy enhancing lesions on the right lobe of the liver in the arterial phase and ill-defined low-density lesions at the same site in the portal phase (Fig. 3B). Nothing remarkable was seen on echocardiography. Bone marrow aspiration and biopsy studies revealed eosinophilic hyperplasia with normal maturation. No blast or FIP1L1/platelet-derived growth factor receptor-alpha (PDGFRA) fusion transcripts were detected, and the results of fluorescence in situ hybridization for BCR/ABL were normal. A chromosomal analysis showed no apparent cytogenetic abnormalities.
The patient was finally diagnosed with HES with involvement of the colon, stomach, duodenum, liver, and lung. Because hypereosinohilia was present for 3 months before his HES diagnosis, hypereosinophic period was shorter than the diagnostic criteria for HES. However, oral corticosteroid therapy (prednisolone, 60 mg/day) was started for HES with severe symptoms caused by multiple organ involvement. Three days later, symptoms such as abdominal pain, bloody diarrhea, and cough were improved dramatically. The patient was discharged, and the steroid dose was slowly tapered. Follow-up blood tests in the outpatient department demonstrated normalization of eosinophil count.
DISCUSSION
Acquired eosinophilia is systemically subclassified into secondary, clonal, and idiopathic types. Secondary eosinophilia originates from parasite infections, allergies, vasculitis, drug use, or lymphoma. Clonal eosinophilia can be distinguished from idiopathic eosinophilia by the presence of histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy.2 HES is a group of disorders with idiopathic eosinophilia that cause damage to multiple organs, and exclusion of both clonal and secondary eosinophilia is required.2-4
HES is defined by the following: 1) persistently elevated eosinophil count (≥1,500/mm3) in the blood for at least 6 months, 2) eosinophilia with no recognizable causes such as parasite infection, cancer, or allergic disease, and 3) symptoms of eosinophilia-mediated organ dysfunction.5 However, a shorter period of hypereosinophilia with symptoms requiring eosinophil-lowering therapy is also acceptable.2
HES with a predominant GI symptom is very rare.6,7 Eosinophilic gastroenteritis (EG), also a rare disease, affects the esophagus, stomach, small bowel, and large bowel.8 EG is diagnosed by the presenting GI symptoms, pathologically eosinophilic infiltration of the GI tract, no eosinophilic involvement of multiple organs outside the GI tract, and no parasite infestation.8 In contrast to EG, HES involves multiple organs and has a progressively fatal course.9 Therefore, careful assessment should be required when evaluating a patient with hyperesinophilia in blood.
Approach for diagnosis requires careful assessment of the peripheral blood smear, bone marrow morphologic features, cytogenic analysis, molecular studies including screening for FIP1L1/PDGFRA, and peripheral blood lymphocyte phenotyping and T-cell receptor gene rearrangement studies.10,11
The primary goal of treatment is to reduce the eosinophilia to <1,500/mm3 and to prevent further damage to major organs.12 Corticosteroid is effective for reducing the eosinophil count, and antineoplastics are useful for slowing eosinophil production.12 Patients who are negative for FIP1L1/PDGFRA fusion should receive a single daily dose of prednisone (60 mg or 1 mg/kg orally) for 1 to 2 weeks, whereas FIP1L1/PDGFRA fusion-positive patients should receive a tyrosine kinase inhibitor (imatinib mesylate) as a first-line therapy.11,13,14 When the eosinophil count is decreased to <1,500/µL, the symptoms are usually relieved. The steroid is gradually tapered to an alternate-day schedule at the lowest dose, which is a median dose of prednisone (10 mg/day).15 Patients who are refractory to glucocorticoid therapy require high-dose glucocorticoid therapy (methylprednisolone, 1 g daily). For cases in which steroid therapy is ineffective, alternative approaches must be considered; these include hydroxyurea, interferon-alpha, anti-interleukin-5, and anti-CD52.16,17 Surgery should be reserved for patients with significant bleeding, perforation, and GI obstruction and for patients who are refractory to medical treatment.16
In our case, early initiation of systemic corticosteriod showed good response to the HES patient with severe hematochezia and chronic cough. Because HES can involve multiple organs and have fatal course, we think eosinophil-lowering therapy should be started as soon as possible to prevent aggressive disease progression and organ dysfunction.
 XML Download
XML Download