

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Niemann-Pick disease type C (NP-C) is a rare autosomal recessive neurovisceral lysosomal lipid storage disorder caused by mutations in NPC1 (95%), NPC2 (4%), or possibly other as yet unidentified genes (1%) [1]. Mutations in either the NPC1 or NPC2 gene lead to cellular deficits, including impaired intracellular lipid trafficking and cholesterol esterification [2]. The estimated incidence of the disease is 1 case in every 120,000 live births, although this is probably an underestimation owing to failure of diagnosis in some cases [3].
NP-C is characterized by visceral, neurological, and psychiatric manifestations that can present alone or in combination. The disorder has a highly heterogeneous clinical presentation at different periods. Consequently, the diagnosis of NP-C may be challenging and the condition may be misdiagnosed. This report describes the case of a patient with NP-C presenting with gait disturbance with spasticity at both ankles, whose condition was initially misdiagnosed as spastic cerebral palsy (CP).
Go to : 
CASE REPORT
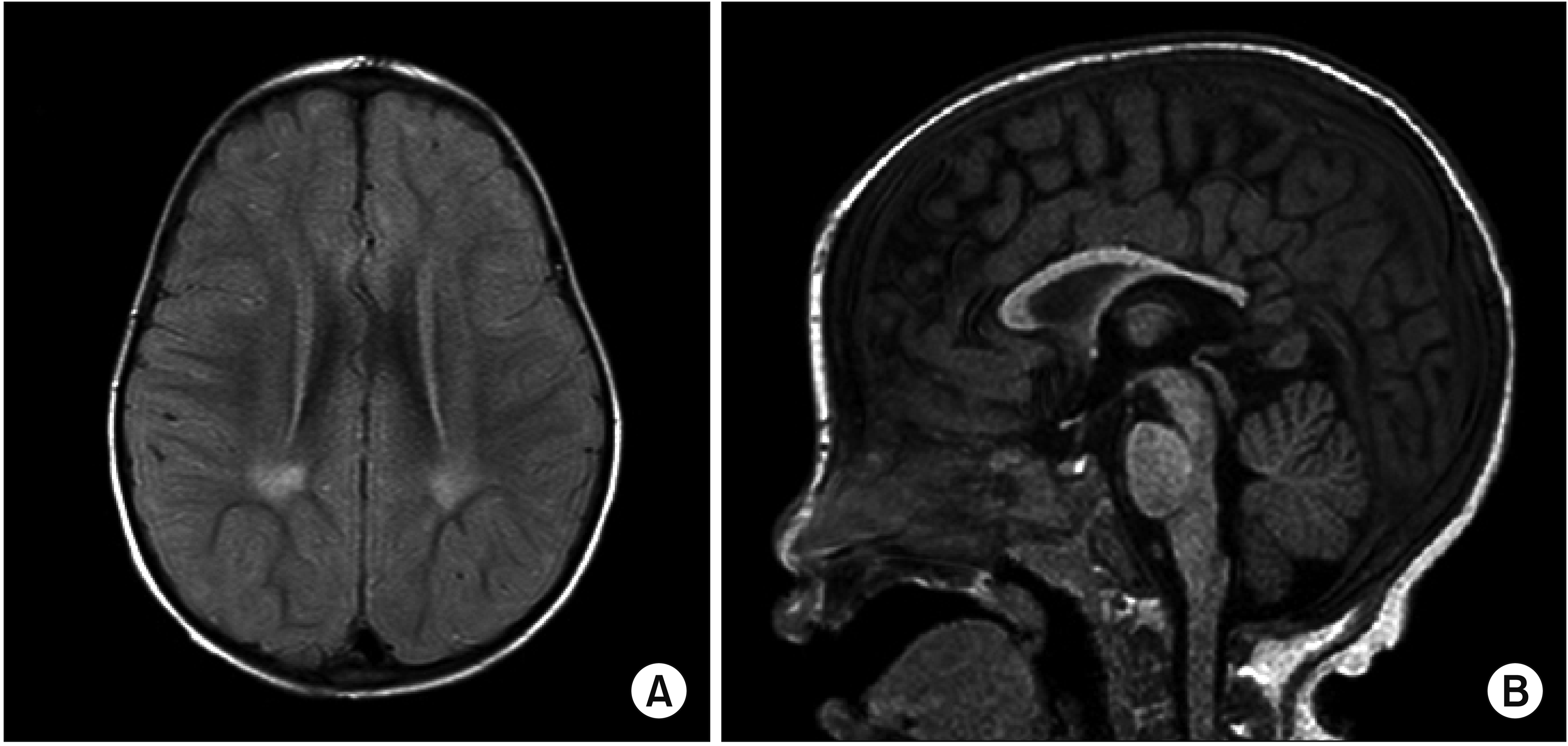
A 27-month-old girl visited the outpatient clinic of the Department of Pediatric Orthopedic Surgery for gait disturbance beginning at the age of 26 months. She showed mild limping with an intoeing gait pattern and spasticity at both ankles. Brain magnetic resonance imaging (MRI) at the age of 29 months revealed findings suspicious of sequelae from a previous insult, such as periventricular leukomalacia (Fig. 1), leading to the diagnosis of CP.
The patient was born at the gestational age of 37+3 weeks, with a weight of 2,035 g (<3%tile), height of 44 cm (3%tile–10%tile), head circumference of 31.5 cm (3%tile–10%tile), and Apgar score of 8>9, through spontaneous delivery. She was the first child, and her family history was not remarkable. Fetal ultrasonography revealed splenomegaly, hepatic congestion, echogenic bowel, and fetal ascites. The ascites resolved after abdomino-amniotic shunt operation in the fetal stage. After birth, she was treated for intrahepatic cholestasis. Her hepatosplenomegaly was continuously observed with ultrasonography. Tandem mass spectrometry screening and amino acid analysis showed nonspecific findings at 1 month.
She visited the outpatient clinic of the Department of Pediatric Rehabilitation Medicine and was admitted at the age of 31 months. Physical examination revealed spasticity in the lower extremities, particularly at the ankles (right<left), which was believed to be a symptom related to mild CP. She had global developmental delay (Table 1) and underwent rehabilitation treatment. Botulinum toxin injection targeting the left equinovarus foot and use of an ankle-foot orthosis helped with her gait pattern.
Neurological symptom deterioration was discovered incidentally via a video of the patient walking before the age of 2 years, which showed better gait pattern than that at the age of 31 months. Therefore, progressive diseases other than CP were considered. However, tests for arylsulfatase A enzyme for metachromatic leukodystrophy, very long-chain fatty acids for adrenoleukodystrophy, and beta-galactosylceramidase for Krabbe disease showed normal findings.
A few weeks later, the patient experienced sudden collapse of the knees when walking, which was first interpreted as an epileptic crisis; however, there was no abnormal finding on electroencephalography. The frequency of knee-collapse events increased, and cataplexy was gradually suspected because knee collapse also occurred when laughing. Furthermore, vertical supranuclear gaze palsy (VSGP) and wide ataxic gait pattern appeared progressively. The NPC1 gene for NP-C showed a mutation in a genetic study, and NP-C was diagnosed.
From the age of 34 months, treatment for NP-C, including oral miglustat and intrathecal and intravenous betacyclodextrin, was initiated and continued. However, her clinical manifestations continued to worsen (Table 1).
The informed consent was waived for this case report.
Go to :
DISCUSSION
Differentiating slowly progressive neurological disorders from CP can be challenging, because these conditions frequently share similar patterns of motor impairment. CP describes a group of permanent disorders of movement and posture that are attributed to nonprogressive disturbance of the developing fetal or infant brain [4]. The key characteristic distinguishing NP-C from CP is the progressive pattern of the disease. Furthermore, some uncommon features of NP-C may help in diagnosing this condition, including visceral, neurological, and psychiatric symptoms.
Visceral symptoms are the most specific symptoms of NP-C [5]. Splenomegaly is the strongest visceral indicator of the disease [1]. Hepatomegaly is observed less frequently than splenomegaly. Prolonged or unexplained neonatal cholestatic jaundice is also a strong visceral indicator of NP-C [5]. The presence of fetal ascites or hydrops fetalis is less frequent [5].
Neurological symptoms are the most common and sensitive indicators of NP-C [5]. VSGP occurs owing to selective vulnerability of neurons at the rostral interstitial nucleus of the medial longitudinal fasciculus and causes deficits in voluntary and reflexive vertical saccades as well as vestibulo-ocular nystagmus [6]. Cataplexy is a sudden episode of muscle weakness with full conscious awareness, triggered by laughing, crying, or the emotion of fear, which can appear at as early as 2 years of age [7]. It is frequently misinterpreted as a fall or as atonic epileptic crises [1]. VSGP and cataplexy are the strongest neurological indicators of NP-C [5]. Slow ataxia is a moderate indicator of NP-C and becomes highly suggestive of NP-C in combination with dystonia of the hands and face [5]. Hypotonia is usually observed in the second year, and spasticity is sometimes observed in the early or late infantile periods. Developmental delay, dysarthria, and dysphagia can also occur [5].
Psychiatric symptoms are the least specific indicators of NP-C [5]. All patients present with progressive cognitive decline, which is a strong indicator of the disease [5]. Psychotic symptoms (hallucination or delusion) are moderate indicators of NP-C [5] and typically appear during adolescence or early adulthood.
The radiographic features of NP-C are nonspecific, among which cerebral (particularly frontal lobe) and cerebellar atrophy [3,8], high T2-weighted signal intensity in the parieto-occipital periventricular white matter [8], deep gray matter and hippocampal atrophy in adultonset NP-C [9], and reduced midbrain-to-pons ratio [10] have been reported. However, the patient with NP-C in this report did not show the above-mentioned findings on brain MRI.
Among the above features of NP-C, the patient in this case report showed hepatosplenomegaly, cataplexy, VSGP, and spasticity; however, interestingly, she did not show symptoms of dystonia, hypotonia, cognitive decline, hallucination, and delusion. Therefore, this case is different from typical NP-C cases.
In conclusion, this report highlights the importance of suspecting other diseases when deterioration of symptoms is detected in a patient with CP-like symptoms, because early treatments for metabolic diseases are important to stabilize or slow disease progression. Therefore, when a patient with CP-like symptoms presents with a deteriorating course and NP-C-specific symptoms, NP-C should be cautiously considered.
Go to :
 XML Download
XML Download