

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
A cerebral aneurysm is an outpouching of a weakened arterial wall with a prevalence that is reported to be 2–4% in the general population [1]. Cerebral aneurysms are usually silent over their lifetime, but they sometimes can be complicated by subarachnoid hemorrhage or mass effect, causing substantial injury to multiple brain areas with a high fatality rate of 35% [2]. The potential for fatal outcomes has led us to investigate the natural history of these lesions and to provide standards for screening and giving prophylactic interventions. In Korea, screening for cerebral aneurysms is recommended for individuals who have 2 or more first-degree relatives with cerebral aneurysm, autosomal dominant polycystic kidney disease (ADPKD), or previous aneurysmal subarachnoid hemorrhage [3]. Screening is not recommended for those with a negative family history and no known risk or genetic factors related to the cerebral aneurysm [4]. The decision regarding whether to treat or not should be determined by considering sufficient patient- and aneurysm-specific factors: age, life expectancy, comorbidity, history of previous subarachnoid hemorrhage, family history, anxiety, aneurysm size, location, multiplicity, morphology, increasing size or morphological change during follow-up, and the assumed risk of the treatment [3].
Despite multidisciplinary discussion according to guideline recommendations, the decision for prophylactic treatment is often difficult in actual practice. Most current knowledge regarding aneurysms that are high-risk for rupture has been derived from limited clinical and aneurysm parameters. Risk factors including hypertension, smoking, and female gender are not enough to explain the interpersonal differences in vulnerability to aneurysm rupture. We are now in search of pathophysiology-based risk markers to better identify high-risk aneurysms. Beyond evaluating final phenotypes shown in angiography, we need to evaluate the early pathological changes that occur during the formation, growth, and rupture of the aneurysms. This review will address the anatomical and embryonic origins of the cerebral artery, the mechanisms of vessel wall stress and degeneration, and markers for high-risk aneurysms, based on new pathophysiological considerations on cerebral aneurysms.
Go to : 
STRUCTURE OF THE CEREBRAL ARTERY
Sound anatomical structure ensures the functional integrity of cerebral arteries; however, its perturbations lead to the development and progression of various cerebrovascular diseases. A cerebral aneurysm and its complications involve structural changes in the arterial wall. A better understanding of the anatomical and embryonic characteristics of cerebral arteries is a basis for pathophysiological consideration of cerebral aneurysms.
Characteristics of cerebral arteries
The cerebral artery, like other systemic arteries is composed of the tunica intima, tunica media, and the adventitia [5]. The tunica intima is the innermost layer and is lined with endothelial cells. The tunica media consists of cellular and matrix components. The adventitia is the abluminal layer of the vessel and is composed of fibroblasts and a collagen-rich extracellular matrix. The internal elastic lamina partitions the tunica intima and tunica media, and the external elastic lamina demarcates the adventitia from the tunica media. The cells and matrix in each layer provide a structural and functional support to maintaining the integrity of the vessel wall. In particular, smooth muscle cells and an extracellular matrix that are oriented perpendicularly to each other in the tunica media confer contractile and regulatory functions, and thus contribute most to the structural support of vessels [6]. The extracellular matrix includes elastin, collagen, proteoglycans, and fibrillin, which are generated and regulated by smooth muscle cells. Extracellular matrices are organized with each other between the lamellar layers of elastin, which confers mechanical strength to the vessel. The turnover of smooth muscle cells and elastin is very low. The lamellar layers of elastin are constructed during the developmental period and show either a slow proliferation or degradation rate of a half-life of approximately 40 years [7]. Therefore, the elastin layer integrity achieved during the developmental period is critical for preserving vascular health over a lifetime.
There are some anatomical differences in the systemic vasculature. Arteries are classified as muscular or elastic according to the composition of the tunica media. The constituents and cellular origins of the cerebral arterial walls vary per vessel site [8,9]. Each arterial site may have a different degree of durability and vulnerability to a variety of pathophysiological signals. The common carotid artery is an elastic artery, and the internal carotid and intracranial arteries are muscular arteries. The intradural and extradural portions of the intracranial arteries have different layer structures. The adventitia in the intradural portion is thinner than that in the extradural one. The collagen component in the adventitia is advantageous for preventing rupture in the face of abrupt pressure changes [10]. Intradural segments also show marked attenuation of elastic fibers. They lack the external elastic lamina that makes the intracranial artery, especially the intradural segment, more vulnerable to aneurysm formation and rupture than other muscular arteries. The posterior vasculature is more capable of arterial remodeling compared to the anterior vasculature [11]. The branching site is more vulnerable to hemodynamic stress because of the deflection and oscillation of blood flow [12], and cerebral aneurysms occur preferentially at arterial bifurcations.
Embryonic origins of cerebral vascular smooth muscle cells
Smooth muscle cells and the extracellular matrix in the tunica media offer structural and functional support to the cerebral artery, and thus the characteristics of these cells are important to maintain a vascular health. Smooth muscle cells act to maintain the structural integrity of mature vessels, which differs depending on the vessel location or the specific segment of the same vessel, which is also related to its distinct embryonic origin [13]. There are two distinct populations that give rise to vascular smooth muscle cells. One is the mesoderm, which forms the dorsal aorta. The other is the neural crest cell population, which generates smooth muscle cells in the beginning portions of the aorta, extracranial arterial trunk, and intracranial arteries [14]. The neural crest cells migrate into the anterior and ventral head, whereas the progenitor cells of mesodermal origin migrate into the dorsal and posterior parts of the head and neck [14]. The vascular trees of two different origins are connected and diverge at the circle of Willis. A key function of smooth muscle cells of neural crest origin is secretion, whereas the mesodermal cells have a contractile function. Smooth muscle cells of neural crest origin produce a higher amount of elastin, but confer a lower contractile function compared to those of mesodermal origin.
Aneurysmal changes can involve multiple vessels of a common embryonic origin when neural crest cells are malpositioned or malfunction. Since the thoracic and abdominal aorta have different embryonic origins, thoracic and abdominal aortic aneurysms have different pathophysiological mechanisms and clinical features [15,16]. Epidemiological observations showed that patients with thoracic aortic aneurysms have a nine-fold higher prevalence of cerebral aneurysms than the general population [17]. It was recently found that ascending aortic aneurysms coexisted more often with aneurysms of the anterior and middle cerebral arteries, whereas abdominal aortic aneurysms occur more often with internal carotid artery aneurysms [18]. As the neural crest cells show high migration and differentiation properties throughout the vessel, heart, and head and neck structures during early embryogenesis [14], perturbations in the development of neural crest cells disrupt the integrity of the vessel walls and other head and neck structures. There have been anecdotal reports showing that cerebral aneurysms are common pathological features of neurocristopathy, such as bicuspid aortic valve [19], congenital heart diseases [20], neurofibromatosis type 1 [21], fibromuscular dysplasia [22]. In line with this concept, patients with multiple, larger aneurysms and rupture tended to have a dilated aortic root [23].
Go to :
PATHOPHYSIOLOGY OF A CEREBRAL ANEURYSM
A cerebral aneurysm accompanies robust structural changes in the vascular wall, as shown in Fig. 1. The pathophysiological components of a cerebral aneurysm give clues to identifying potentially susceptible patients. Although the exact pathogenesis of cerebral aneurysm formation, growth, and rupture remains to be established, extracellular matrix defects and degeneration, hemodynamic stress, and inflammatory responses have been suggested as key components leading to structural fragility in the arterial wall. Each of the components is closely interconnected. The perturbations of structural component are specified in Table 1.
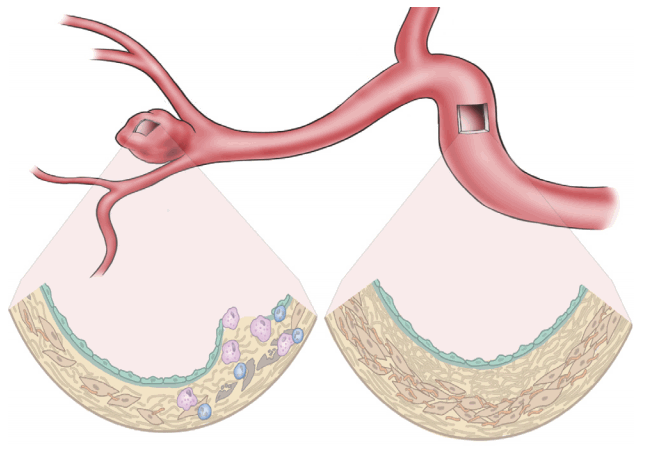 | Fig. 1.Structural alteration of a cerebral aneurysm. Intracranial arteries are normally composed of firm layers, including endothelial cell, internal elastic lamina, smooth muscle cells, extracellular matrices, and adventitia. Meanwhile, aneurysmal changes result from perturbations of one or more of these components.
|
Table 1.
Pathophysiological factors involved in aneurysm formation
VCAM-1, vascular cellular adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; TNFα, tumor necrosis factor-α; MCP-1, monocyte chemoattractant protein 1; IL, interleukin; COX, cyclooxygenase; PGE2, prostaglandin E2; NO, nitric oxide; TLR, toll-like receptor; MMP, matrix metalloproteinases.
![]()
Extracellular matrix defect and degradation
The extracellular matrix is a dynamic structure that is continuously undergoing a remodeling process by interacting with vascular cells. Given the secretory function of smooth muscle cells in the cerebral artery rather than contractile function, the mechanical strength of the large arteries primarily depends on the cross-linking of elastin and collagen. The longevity of elastin generated in early embryogenesis is similar to the human life span, and rarely experiences a wear and tear process. Once extracellular matrix defects or degradation take place, the disease course may not be restored. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases that are produced by smooth muscle cells and inflammatory cells mediate the process of extracellular matrix degradation and remodeling. An imbalance between MMPs and their inhibitors contributes to initiation and progression of cerebral aneurysms.
Lysyl oxidases (LOX) catalyzes the critical step in cross-linking elastin and collagen [24]. LOX requires one tightly bound copper ion for its active role. As dietary copper amount directly affects LOX activity [25], copper deficiency during the developmental period may attenuate LOX activity and weaken vessel wall integrity leading to the development of aneurysms in adulthood. It was recently demonstrated that copper deficiency in mice during the developmental period caused complex vascular wall abnormalities involving thoracic aortic aneurysms and cerebral aneurysms [26]. The thoracic aortae were dilated with disorganized elastic fibers, and the fusiform and saccular aneurysms were noted in the surviving mice. Since copper deficiency often occurs during infancy in cases of cow’s milk feeding or infant formula with low copper content [27,28], the infancy environment and food habits may affect aneurysm prevalence and outcome. Clinically, a variety of extracellular matrix defect have been detected in patients with connective tissue diseases such as osteogenesis imperfecta, vascular Ehlers-Danlos syndrome, and Marfan syndrome, which are commonly associated with cerebral aneurysms.
Hemodynamic stress
Cerebral aneurysms preferentially occur at the anterior communicating artery, the posterior communicating artery, the middle cerebral artery bifurcation, and the basilar artery bifurcation where local shear stress is greatest on the arterial wall [12,29]. Blood flow at the arterial junctions, the bifurcations with wider bifurcation angles, or abrupt vascular angles are the most turbulent, and the shear stresses in these areas are the greatest. High wall shear stress induces endothelial cell damage, smooth muscle cell degeneration, and media thinning. Hemodynamic forces also cause endothelial cells and smooth muscle cells to release MMP-2 and MMP-9 and subsequently to degrade the extracellular matrix, resulting in aneurysm formation. The magnitude of local shear stress is well correlated with the degree of internal elastic lamina loss, medial degeneration, and arterial bulging [30]. Shear stress is certainly a strong trigger for developing aneurysms in individuals that are predisposed to them. In this regard, flow diverters to attenuate shear stress have been recently applied to decrease the risk of aneurysm growth and rupture [31]. On the other hand, recent investigations by computational fluid dynamics models support the differential role of hemodynamics on development and rupture of cerebral aneurysms. While a high wall shear stress promotes formation of aneurysms, low stress has been associated with aneurysm rupture [32]. Moreover, the wall shear stress is significantly lower at the rupture point that sac [33], and pooled analyses show a decreased wall shear stress could predict aneurysm rupture [34].
Smoking is associated with a higher prevalence of cerebral aneurysms and a higher risk of rupture. The mechanisms by which smoking results in cerebral aneurysm formation and rupture are suggested to be elevated wall shear stress due to increased blood volume and viscosity and nicotine-induced vasoconstriction [35]. Extracranial carotid disease may increase shear stress hemodynamically on the contralateral distal vascular bed by increasing the blood volume. According to this hypothesis, contralateral carotid artery ligation methods have been used to create experimental cerebral aneurysms; however, inconsistent and inefficient modeling has resulted. When we investigated the relationship between extracranial carotid disease and characteristics of cerebral aneurysms, aneurysms were evenly distributed irrespective of stenosis laterality [36]. The rate of aneurysmal growth and rupture in patients with significant extracranial carotid artery stenosis was also low [36]. Wall shear stress at the aneurysm site appears to be influenced more by adjacent geometrical changes rather than by remote input [37].
Inflammatory response
Cerebral aneurysms have been histologically characterized by acute and chronic inflammation and medial wall degeneration [38,39]. Numerous immunologic factors have been identified as initiators and inducers of cerebral aneurysm development. High hemodynamic stress alters endothelial function and induces an infiltration of inflammatory cells and activation of downstream cascades in the tunica media [40,41]. Monocyte chemoattractant protein 1, NF-κB, angiotensin II, prostaglandin E2, and prostaglandin E receptor subtype 2 amplify this response and lead to phenotypic switching in smooth muscle cells [39-43]. Phenotypically-switched smooth muscle cells evoke pro-inflammatory and pro-matrix remodeling and undergo apoptotic cell death. The MMP produced from monocytes/macrophages and smooth muscle cells plays a role in extracellular matrix degradation and vascular remodeling. Interleukin (IL) 1β, IL6, tumor necrosis factor-α, TLR4, Fas, nitric oxide, and complement have been suggested to function as key molecules in the propagation of immune responses [39,44]. High concentrations of cytokines and chemokines have also been detected in the human plasma associated with cerebral aneurysms [45].
These inflammatory responses work in concert to weaken the vessel wall, thereby developing aneurysmal dilatation and eventually rupture. In our experimental aneurysm model, angiotensin II infusions to rats exposed to copper deficiency during the early developmental stage showed a higher rate of complicated cerebral aneurysms with rupture, which suggests that angiotensin II may increase the medial degeneration possibly via upregulation of inflammatory cascades [26,46]. Vasculitis is an inflammatory disease of the cerebral vessels that is mediated by infiltration of infectious pathogens or inflammatory cells into the vessel wall. Infection, autoimmune disease, or trauma have been associated with multiple aneurysms, which could be reversed by anti-inflammatory drugs. Pharmacological approaches targeting the inflammatory cascade may be effective in the treatment of cerebral aneurysms [47].
Go to :
RISK FACTORS AND BIOMARKERS ASSOCIATED WITH FORMATION AND RUPTURE OF CEREBRAL ANEURYSMS
Aneurysmal subarachnoid hemorrhage has a bimodal age distribution pattern. Advancing age and vascular risk factors explain the older-age peak, and intrinsic wall defects contribute to the younger-age peak. Cerebral aneurysms are a complex heterogeneous disease with a variety of outcomes and susceptibilities to growth and rupture. Growth or de novo formation of cerebral aneurysms are important morphological markers for instability of an unruptured aneurysm [48]. We need to stratify the risk of cerebral aneurysms based on patient and/or aneurysm-specific factors underlying the instability of cerebral aneurysms.
Conventional risk factors for rupture
Aneurysmal factors include size, location, and morphology, while patient factors include female sex, current smoking, alcohol consumption, hypertension, history of subarachnoid hemorrhage, and positive family history [1,2]. Aneurysms larger than 10 mm have a 1% risk of rupture per year. Aneurysms of the anterior communicating artery rupture more easily in smaller sizes than those in other locations [49,50]. Although larger aneurysms usually have a higher risk of rupture, ruptured aneurysms are usually small. Most aneurysms develop over a relatively short period of time during which they are at highest risk of rupture, even though small. If the size increase or morphological changes occur within a short time period, the likelihood of rupture increases [51]. Multiple aneurysms are prevalent in pediatric and young adult patients, and are prone to recurrent subarachnoid hemorrhage [52]. Morphological changes suggestive of a higher risk of rupture include the presence of daughter sac, a high dome/neck ratio, and multilobularity [53]. With these aneurysm- and patient-specific factors, risk scoring system such as PHASES has also been suggested to identify patients at high risk for rupture [54]. Nevertheless, a substantial portion of patients with cerebral aneurysms have no risk factors. Clues for predicting who will develop a cerebral aneurysm and in which artery are still lacking. Outcomes that each aneurysm can exhibit are unpredictable.
Innate risk factors
Some aneurysm cases display eccentric features, including large size, multiplicity, rupture presentation as small aneurysm, and combined vasculopathy in the systemic vasculature (Fig. 2). An intrinsic vascular wall deformity may be a major contributor to this group. The contribution of an intrinsic wall deformity to the pathophysiology of a cerebral aneurysm has been supported by genetic, experimental, and clinical studies. Genetic disorders with a variety of phenotypes have been reported to also present with cerebral aneurysms (Table 2) [55,56]. Although the link between the involved gene and the aneurysm phenotype is not fully understood, there are likely some common genetic predispositions to the main clinical features and aneurysm. ADPKD involves defects of PKD1 and PKD2 genes, which are associated with endothelial barrier function. About 20–40% of ADPKD cases have cerebral aneurysms, and 10–30% of those have multiple aneurysms [57]. The risk for de novo aneurysm and rupture in patients with ADPKD is also higher than in the general population. In fibromuscular dysplasia, the prevalence of cerebral aneurysms is reported to be about 13%, which is about six times higher than that of the general population [58].
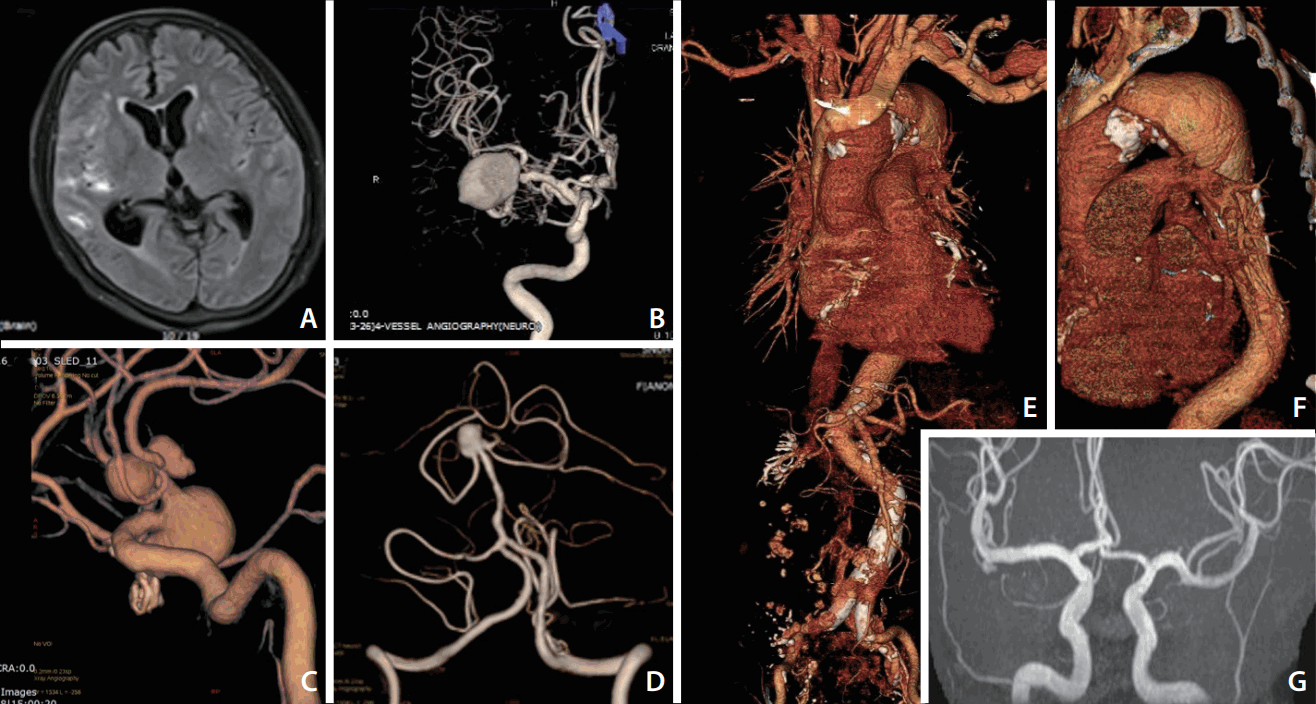 | Fig. 2.Cerebral aneurysms with eccentric features associated with intrinsic vessel wall deformities A 55-year old woman was admitted because of an aneurysmal subarachnoid hemorrhage (SAH) in the right sylvian fissure (A). A large aneurysm in the right middle cerebral artery (MCA) was considered as a culprit lesion of SAH (B). However, multiple, large aneurysms were also noted in the anterior communicating and anterior cerebral arteries (C), and basilar artery (D). The second patient was a 53-year old woman complaining of anterior chest discomfort. Computed tomography angiography showed a large aneurysm involving the descending thoracic aorta (E, F). An intracranial aneurysm was also detected in the right MCA bifurcation from the screening magnetic resonance angiography (G).
|
Table 2.
Hereditary disorders with susceptibility to an intracranial aneurysm
![]()
Cerebral aneurysms are also prevalent in patients with a bicuspid aortic valve and coarctation of the aorta [19,59]. Since the heart outflow tracts and the cerebral artery share neural crest cell origins and pathological changes, combinations of these congenital heart diseases and the development of cerebral aneurysms are considered to be phenotypes of neurocristopathy [60]. It was also found that dilated aortic roots with no overt heart disease is associated with eccentric aneurysm features, such as large size, multiple lesions, or presentation with rupture at a young age [23]. The concept of intrinsic wall deformity as a main contributor to a cerebral aneurysm has been recently confirmed in the copper-deficient model during the developmental period, which displayed an aortic and cerebral aneurysm [26]. A multifactorial genetic predisposition would more strongly underlie the pathogenesis of a cerebral aneurysm formation than a single genetic disorder. Significant genes associated with intrinsic wall deformities have been determined from whole exome sequencing and genome-wide association studies, but the findings were inconsistent and conflicting [56]. Disease heterogeneity, different levels of genetic burden per stage, and gene-environment interaction would explain the genetic complexities of the cerebral aneurysm. It is supposed that cerebral aneurysms result from the impact of various acquired factors imposed on a genetically determined defect of the vessel wall.
Acquired risk factors
Acquired inflammatory conditions such as atherosclerosis, infection, and trauma can weaken the arterial wall, resulting in cerebral aneurysm formation. The development of cerebral aneurysms at relatively older ages is associated with age-related risk factors and accumulating atherosclerosis during the aging process. Acquired wall degeneration may contribute to aneurysm formation at atherosclerosis-prone sites. Our recent study showed that abdominal aortic aneurysms are more frequently associated with distal internal carotid artery aneurysms [18]. Given that an abdominal aortic aneurysm is caused by atherosclerosis, and that the distal internal carotid artery has more atherosclerotic stenosis than other sites, it is natural that atherosclerosis-related aneurysms would show site-specific patterns.
There are other acquired risk factors to be proven as aneurysm-inducing factors. Smoking induces the inflammatory response in the cerebral vessel and weakens the wall [61]. A recent study emphasized that the intensity and duration of smoking is well correlated with the risk of cerebral aneurysm rupture [35]. Moreover, the cumulative effect of smoking on the risk of aneurysmal subarachnoid hemorrhage was not reversed with smoking cessation [35]. Sex hormones may contribute to the acquired arterial wall weakness. Women are more susceptible to a cerebral aneurysm and occurrences of multiple aneurysms. Estrogen regulates the collagen metabolism, inflammatory mediators, MMP release, and vascular tone [62-64]. Women undergo different reproductive and hormonal phases in menarche, menopause, oophorectomy, and hormone replacement therapy over their lifetime. Thus, various degrees of estrogen exposure may lead women to have differing susceptibilities to cerebral aneurysms. Sex-related differences in bone health may also support the link between sex hormones and extracellular matrix degeneration [65]. Osteoporosis shows a female predominance, and bone mineral density reflects a cumulative estrogen exposure [66]. Our recent study showed that decreased bone mineral density is associated with cerebral aneurysms, large size aneurysms, and multiplicity of aneurysms [67]. We need more detailed evaluations about sex-specific factors to clarify the causes of these sex-related differences.
Cerebral aneurysms are detected in about one-third of cases of arterial dissection. When the dissection involves the intradural portion of the cerebral vessels, subarachnoid hemorrhage can occur. Since intracranial arteries have no external elastic lamina and the attenuated media has a paucity of elastic fibers, intracranial arterial dissection is more vulnerable to aneurysm formation or rupture than an extracranial one. Although the risk factors and predisposing conditions for cerebral artery dissection are not clear, acquired wall degeneration by trauma seems to mediate this co-occurrence.
Systemic risk markers for aneurysm development and rupture
A significant proportion of patients with a cerebral aneurysm have intrinsic vascular wall deformities, which is associated with a high risk of growth, de novo formation, and rupture of aneurysms. Therefore, patients with unruptured cerebral aneurysms need genetic and clinical investigations into the underlying vessel wall status. As previously mentioned, intrinsic cerebral arteriopathy can display different degrees of aortopathy with no overt cardiac diseases. A common echocardiographic marker, aortic root dimension could be applied to identify intrinsic vascular wall defects [23]. Meanwhile, other echocardiographic markers of the atrial and ventricular wall structures derived from cardiac mesenchyme did not play any role in identifying high-risk aneurysms. In our study, patients with larger aneurysms showed a tendency to be shorter [23]. A negative correlation between height and aneurysm size provided additional systemic evidence of the developmental problems associated with cerebral aneurysm formation. Since neural crest cells differentiate into the skeletal system and regulate growth-related hormones [68], the alteration in neural crest development may underlie a biological process that determines the height and development of a cerebral aneurysm. The extracellular matrix components are widely distributed throughout the body from the vessel walls to the bones. Thus, alteration in matrix properties can affect bone and vascular health concomitantly [69]. Thus, the measurement of bone mineral density in patients with cerebral aneurysms, or vice versa, may be clinically informative.
Go to :
CONCLUSION
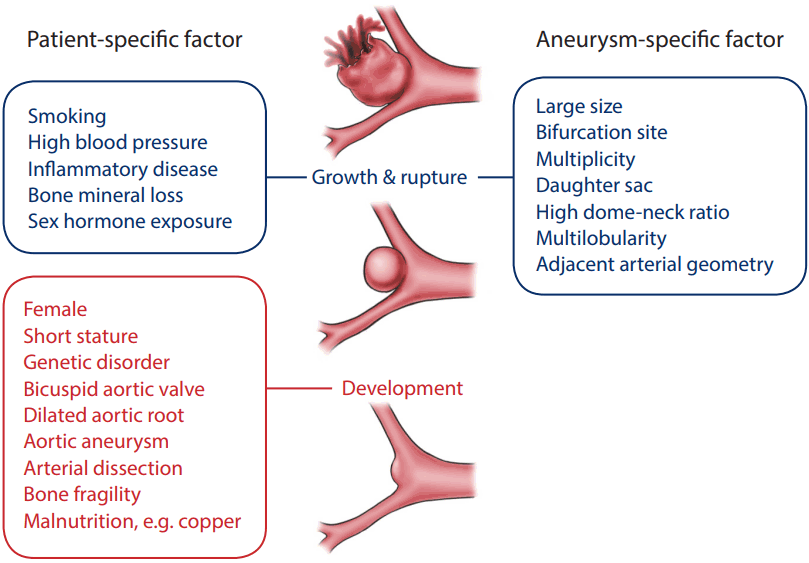
Cerebral aneurysms are asymptomatic and may be found incidentally in cerebral angiographies performed in neurology clinics or in health screening settings. It will be important to decide whether or not to treat them, and how to continue with long-term follow-up in these patients. Close imaging surveillance for morphological changes in the aneurysm or preventive treatment should be provided for some patients with a profile that exhibits a high risk of rupture. However, the majority of cerebral aneurysms require no imaging follow-up or prophylactic treatment. Aneurysms may harbor intrinsic vessel wall deformities or acquired vessel wall degeneration during their development, and they may have variable outcomes according to the different pathophysiological signals. Patient- or aneurysm-derived factors can help to stratify the risk of progression or rupture. Enriched clinical observations and advanced imaging and histological data have enhanced our knowledge of the pathophysiology of cerebral aneurysms and provided new patient- and/or aneurysm-specific factors for consideration (Fig. 3). Future multi-factorial evaluations with genetic, imaging, and laboratory tests would offer the opportunity to better identify aneurysms that have a high risk of rupture.
Go to :
 XML Download
XML Download