

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Distal 5q deletions are rare, with only 12 reported cases of involving the 5q33-qtel region [1]. The chromosome 5q34-q35.2 region carried the susceptibility gene for type 2 diabetes [2].
The various manifestations of distal 5q deletion include congenital cardiac anomalies, craniofacial dysmorphism, intellectual disability, and developmental delay [3]. In addition to common features associated with the distal 5q deletion, our patient manifested a unique clinical phenotype that included torticollis and limited range of motion due to multiple bone anomalies in the cervical spine. Few studies have reported the clinical consequences of musculoskeletal abnormalities involving the 5q deletion. Mild digital abnormalities such as camptodactyly, syndactyly [4], and parietal foramina were observed in a few cases [5]. However, spinal bone abnormalities have not been reported.
We report a female proband carrying a de novo 5q34-q35.2 deletion breakpoint detected by fluorescence in situ hybridization (FISH), and review the unique skeletal phenotype and analyze the possible related genotype.
CASE REPORT
A 4-year-old girl visited our clinic to participate in a rehabilitation program for developmental delay. Because of intrauterine growth restriction, she was born at 39 weeks with a low birth weight of 2,000 g (0–5th percentile). She was the first child with a non-contributory family history. She underwent surgery for a cardiac septal defect at 2 months and was hospitalized due to feeding and respiratory disorders for 7 months. Tube feeding was performed until 8 months, and oral feeding was started at 24 months with persistent signs of aspiration when drinking liquids.
Physical examination revealed dysmorphic features including microphthalmos and an artificial eye along with bilateral hearing aids. The motor ability of both upper and lower extremities was generally fair except that both hip extensors were trace and plantar flexors were poor. The developmental milestones were markedly delayed. Head control was recognized at 12 months, while creeping and crawling were observed at 3 and 4 years, respectively. The patient underwent physical therapy starting at 28 months. She was able to maintain a sitting position without a manual assist, but could not stand up and walk at the age of 4 years. Chromosome analyses revealed 46,XX at the 550 band level. Genetic testing results for Prader-Willi syndrome, Angelman syndrome, and spinal muscular atrophy proved negative. However, a chromosome 5q34-5q35.2 micro-interstitial deletion was diagnosed by FISH before the patient visited our hospital.
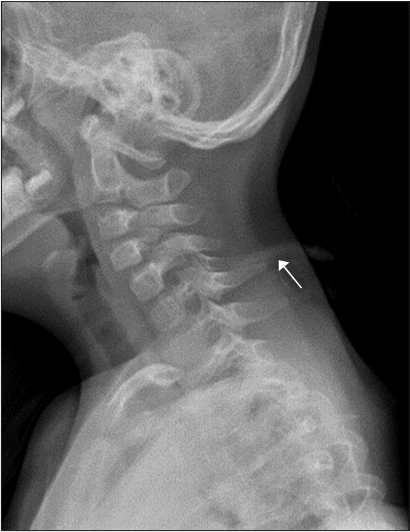
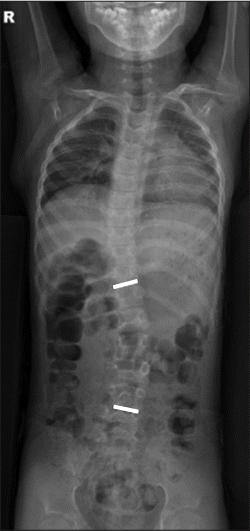
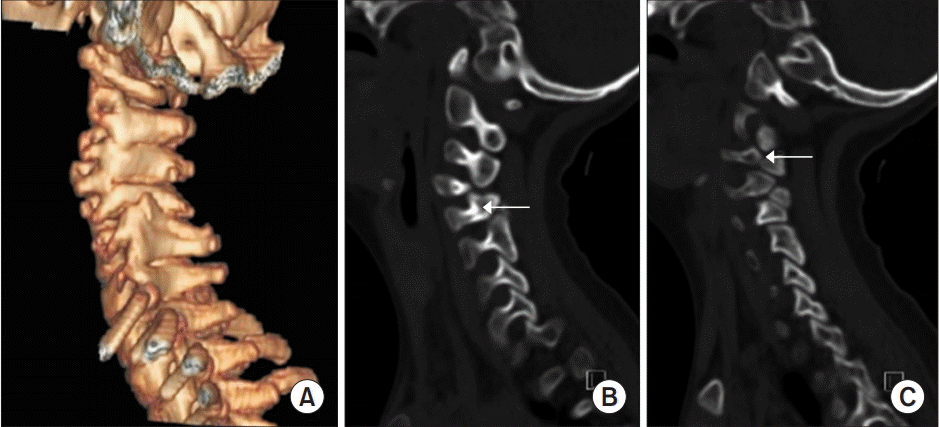
The patient showed a persistent head tilt, which was initially considered a symptom of hypotonia. However, she also had a limited range of head rotation. Therefore, radiographic analysis of the spine was performed. Cervical plain radiographs showed abnormal bony structures (Fig. 1). Whole-spine plain radiographs showed thoracolumbar scoliosis (Fig. 2). Non-contrast-enhanced, threedimensional computed tomography of the cervical spine (Fig. 3A) revealed a bone cleft in right pars interarticularis, a bone defect in both lamina of C5, and agenesis of the right articular process of C5, which was associated with bony fusion of C4–C5 (Fig. 3B). Subluxation of the craniocervical joints was also noticed. An additional focal bone defect in the right transverse foramen induced inferior bony protrusion of the right transverse process and right foramina stenosis at C2–C3 (Fig. 3C). Brain magnetic resonance imaging indicated reduced white matter volume and dilated occipital horns of the lateral ventricles or colpocephaly.
A developmental evaluation using the Bayley Scales of Infant Development administered at 41 months suggested cognitive, receptive, and expressive communication, fine and gross motor skill equivalents of 5, 2, 3, 5, and 7 months, respectively. The total score of the Gross Motor Function Measure-88 at 41 months was 29%. The patient’s Gross Motor Function Classification System level was 4. Her speech was limited to simple vocalization without meaning. The Denver Development Screening Test administered at 5 years revealed personal-social, fine-motor adaptive, gross-motor, and language skill equivalents of 5, 6, 5, and 4 months, respectively.
DISCUSSION
We report cervical spine abnormalities associated with an interstitial deletion in the long arm of chromosome 5, locus 5q34-5q35.2. Several deformities of the upper and middle cervical spine were observed in this patient including a bone defect, increased instability of the craniocervical joints, an abnormal posture, and limited range of motion. This phenotype has not been previously described with the 5q deletion. The phenotype was attributed to the gene corresponding to the deleted region. A total of 61 known genes are registered in the Online Mendelian Inheritance in Man (OMIM) database in this deleted region. However, only NKX2–5, SH3PXD2B, and MSX2 are morbid genes related to a developmental disorder, based on searches of the Developmental Disorders Genotype-to-Phenotype (DDG2P) database. The NKX2–5 (OMIM #600584), which is located at the junction of bands 5q34–35.1, is related to congenital heart disease including atrial septal, ventricular septal, and conduction defects [4]. MSX2 (OMIM #123101), which is located at 5q35.2, affects cell growth, particularly in the membranous part of bone [5]. Aftimos et al. [5] suggested that haploinsufficiency of MSX2 is associated with parietal foramina. By contrast, studies of the overexpression of MSX2 gene reported that trisomy of the 5q region contributed to craniosynostosis [6] and distal 5q duplication encompassing MSX2 resulted in bilateral radial agenesis with absent thumbs [7].
MSX2 is an important developmental regulator underlying limb, craniofacial, and ectoderm formation in vertebrates [7]. Our patient carried a micro-interstitial deletion in the distal 5q segment encompassing MSX2. Based on a review of chromosome 5q patients, the MSX2 gene within the 5q34-q35.2 region may trigger cervical spine abnormalities.
Cervical spine abnormalities have also been reported in association with other genetic syndromes including mutations of genes located on chromosomes 22q11.2 and 3p [8,9]. The prevalence of developmental cervical variation in 22q11.2 deletion syndrome is nearly 100%, in addition to scoliosis and limb malformations. Therefore, evaluations for screening of various skeletal anomalies are recommended in this genetic disorder [10].
Our patient had a micro-interstitial deletion involving 5q34-q35.2, harboring the MSX2 gene. The patient manifested features that included cardiac septal defects, feeding difficulty, scoliosis, cervical spine malformations, and global developmental delay including both motor and language skills. Several cases diagnosed with distal 5q deletions have been reported, but to the best of our knowledge, this is the first report of a de novo 5p34-q35.2 interstitial deletion associated with a cervical spine anomaly. Molecular studies are required to identify the genotype-phenotype correlation of this deletion and to confirm the effects of MSX2 expression.
 XML Download
XML Download