

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Developmental delay (DD) and intellectual disability (ID) are major neuropsychological disorders in children, affecting 1%–3% of the general population. Of DD/ID cases, 17.4%–47.1% can be explained by genetic factors [1].
Recently, diagnostic exome sequencing (DES) has been introduced to analyze the underlying cause of unexplained DD/ID with the discovery of a novel disease gene. Heterozygous de novo mutation of the histone acetyltransferase encoding gene KAT6B has been recognized to cause a syndrome with congenital anomalies and intellectual disability, namely Say-Barber-Biesecker-Young-Simpson syndrome (SBBYSS) [2].
Here, we present a case of SBBYSS in three generations of a Korean family affected with a missense mutation in KAT6B, c.2292C>T p.(His767Tyr) identified by DES. This is the first inherited KAT6B missense mutation reported worldwide.
CASE REPORT
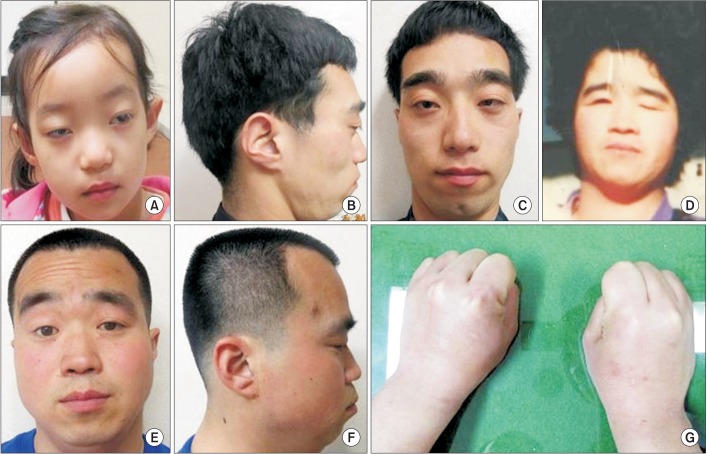
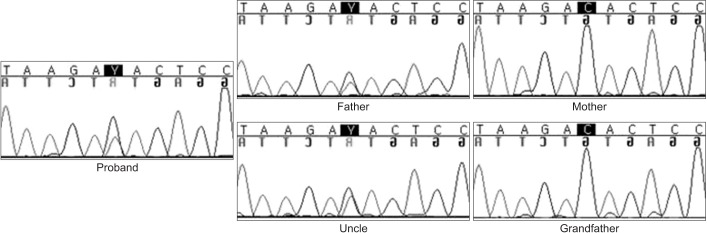
The 3-year-old girl visited the department of rehabilitation medicine for evaluation due to developmental delay. She was the first born child of unrelated nonconsanguineous parents. At birth, mother and father were 23 and 30 years old, respectively. She was born in the 40th week of gestation after an uncomplicated pregnancy. Her birth weight was 3,500 g (50th–75th percentile). At the age 2, bilateral blepharophimosis and strabismus were noticed during an ophthalmologic evaluation. She sat at 10 months and walked at 18 months. Speech development was also slightly delayed. She started to say few simple words at 2 years, and was able to make phrases with 2–3 words at 3 years. Her social and emotional development declined, and she was described as shy, having little interaction with other people. On physical examination at the age of 3, her height was 90 cm (25th–50th percentile), weight 12 kg (below 25th percentile) and her occipitalfrontal circumference was 46 cm (below 3rd percentile). She had an immobile face, blepharophimosis, ptosis, a broad and flat nasal bridge, and a low set of large protruding ears (Fig. 1). Further investigations, routine laboratory tests (including thyroid function tests, brain magnetic resonance imaging, electroencephalogram and joint X-rays) did not reveal any abnormality. During the examination, we noticed that her father also had a similar dysmorphic face. He had a contracture deformity in his metacarpophalangeal joints that prevented him from making a tight fist (Fig. 1). He had graduated from college and was working in a manufacturing job as a simple laborer. According to the Korean–Wechsler Adult Intelligence Scale-IV, his intellectual quotient was 66, indicating intellectual disability. The pedigree acquired from the patient revealed that the patient's uncle was also diagnosed as moderately mentally retarded. Also, her deceased grandmother who was severely mentally retarded (Fig. 2). Given the family history, we arranged a chromosomal microarray for the patient that yielded a normal result. DES was performed on the subject, followed by Sanger sequencing of the family trio (proband and both parents), identified KAT6B missense mutation in c.2292C>T p.(His767Tyr) in the patient and her father. To better understand the relationship between the KAT6B variant and the familiar inheritance pattern, additional familial testing included the grandfather and uncle. The uncle was found to carry the same KAT6B variant (Fig. 3). Considering these results and family history, the patient's grandmother was deemed an obligatory heterozygote. The variant was predicted to be deleterious in silico analysis using Sorting Intolerant from Tolerant (SIFT) with a score of 0.01 (resulting as damaging), Polymorphism Phenotyping v2 (PolyPhen-2) with a score of 0.87 (resulting as possibly damaging), and MutationTaster resulting as disease causing (prob: 0.999999996930964).
They were diagnosed as SBBYSS. Although a few cases with KAT6B have been reported worldwide, this is the first confirmed family inherited mutation of the KAT6B variant to date.
DISCUSSION
SBBYSS is very rare syndrome associated with the KAT6B mutation. Clinical findings include a distinctive facial appearance with severe blepharophimosis, an immobile mask-like face, a bulbous nasal tip, and a small mouth with a thin upper lip, usually accompanied by mental retardation, delayed motor milestones, and significantly impaired speech [3].
The KAT6B gene is located on chromosome 10q22.2 and encodes lysine acetyltransferase 6B, a part of the histone H3 acetyltransferase complex in adult neural stem cells. Little is known about the function of the histone acetyltransferase, but it appears to regulate genes that are important for early development, including development of the skeleton and nervous system [4]. Recently, similar KAT6B mutations were identified in subjects with genitopatellar syndrome (GPS). GPS shows more severe conditions than in SBBYSS with characteristic features including structural brain defects (agenesis of corpus callosum), large joint contractures, genital anomalies, hydronephrosis with multiple renal cysts, and absent patella [5].
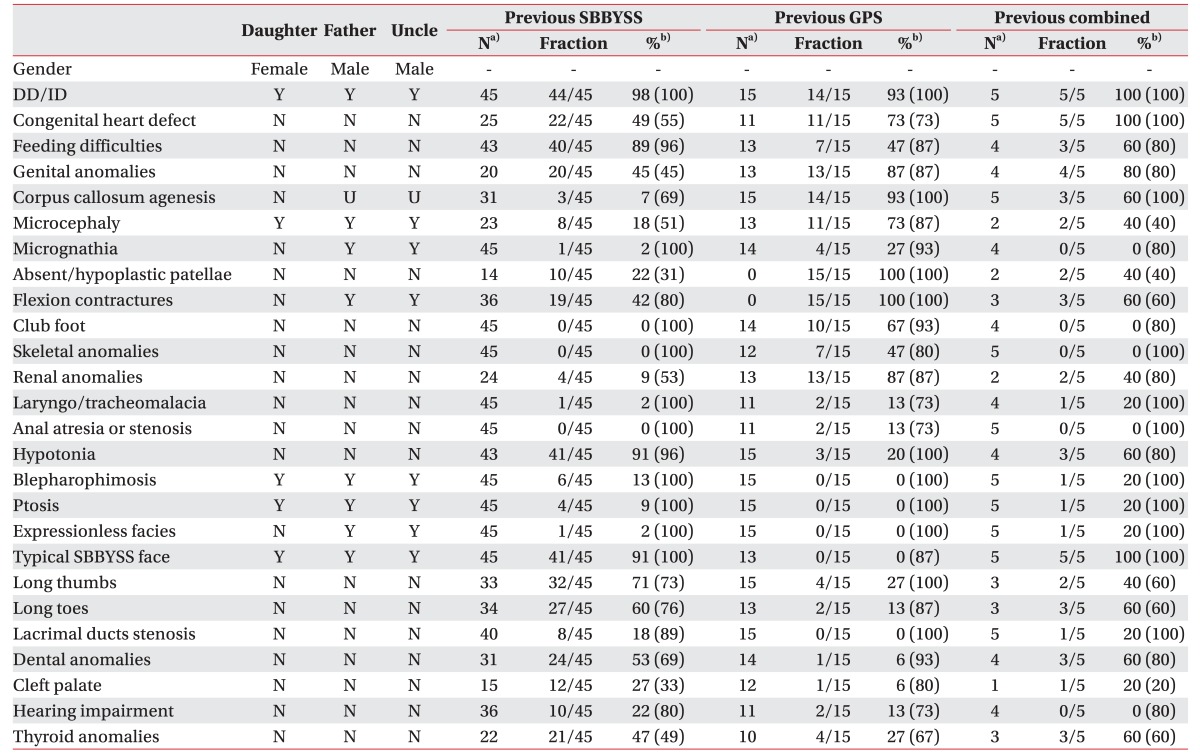
Until now, 45 SBBYSS, 15 GPS patients, and 5 additional patients with combined phenotypes between the two syndromes have been reported with KAT6B mutation [235678]. Of those 65 KAT6B mutations, 53 mutations were located in exon 18. This is the last KAT6B exon responsible for encoding acidic (A, Asp/Glu-rich) and transcriptional activation (TA, Ser/Met-rich) domains of the KAT6B [6]. Gannon et al. [7] suggested that SBBYSS mutations occurred more distally in exon 18 preserving the whole acidic domain but lacking the transcriptional activation domain that led to loss-of-function mutation. While GPS mutations were located more proximally in the same exon, losing both domains led to gain-of-function mutation. However recent studies identified mutations more proximal in exon 15, 16, 17 with the typical features of SBBYSS [78]. The mutation occurred in exon 11 in our patient. Premature termination codon located proximal to the last exon is known to cause nonsense-mediated RNA decay [9] resulting in haploinsufficiency of KAT6B with mild SBBYSS phenotype (Table 1). Clayton-Smith et al. [2] also reported a patient with KAT6B variants seen in the proximal part of exon 3 with mild SBBYSS phenotype. Our report confirms the previous observations that position of the mutation in the gene correlates with the phenotypes of the patients.
All KAT6B variants identified so far have had heterozygous truncating de novo with no familiar recurrence [235678]. This report demonstrates a family with an inherited KAT6B missense mutation as opposed to the de novo mutations seen in SBBYSS and GPS.
In this report, DES was crucial for a correct diagnosis. DES has proven to be a powerful tool with a 25% diagnostic yield for the identification of pathogenic variants leading to Mendelian disorders in DD and ID [10]. DES can detect single-nucleotide substitutions, and insertions or deletions, of 8 to 10 nucleotides or smaller. It is suggested that DES, together with array-based comparative genomic hybridization array, is a first-tier diagnostic tool to evaluate unexplained DD and ID. Patients with DD and ID often undergo a lengthy, time-consuming process for a definitive diagnosis, commonly known as a ‘diagnostic odyssey.’ Establishing an exact diagnosis in patients with DD and ID is important because it brings comfort to the patient's family, enables prediction of the disease's clinical course with relative certainty, allows formulation of an appropriate rehabilitation plan, and maintains general health. It also assists in accurately determining the risk of recurrence in subsequent pregnancies. Physiatrists are likely to be the first to encounter these patients. Therefore, to ensure a correct diagnosis, a careful physical examination that gathers detailed information on family history, and an appropriate genetic analysis, should be considered for patients with unexplained DD or ID.
To the best of our knowledge, this is the first molecular genetic analysis of a SBBYSS patient from the Korean population, and the first three generation inherited KAT6B missense mutation seen worldwide. This report broadens the phenotypic spectrum. Furthermore, the possibility of gonadal mosaicism must be considered to explain the rare familial recurrence of this condition. We also showed that DES can be useful tool to identify causative pathogenic variants in patients with DD and ID.

 XML Download
XML Download