

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Motor neuron diseases (MNDs) are rare neurological disorders caused by selective degeneration of motor neurons that manifest as progressive weakness of various muscles. MNDs are classified according to area affected by degeneration: upper motor neurons (UMNs), lower motor neurons (LMNs), or both. Loss of UMNs results in slowness of movement, incoordination, hyperreflexia, and spasticity. Consequences of LMN degeneration include weakness, usually accompanied by atrophy and fasciculation, caused by loss of LMNs in the brainstem and spinal cord [12].
The initial signs and symptoms of MNDs vary depending on the patient, but typical symptoms include motor weakness and a combination of wasting of muscles, dysphagia, and dyspnea. Most patients with MNDs display respiratory failure; however, it is extremely rare to find any patient who presents with only respiratory dysfunction as the initial symptom [3456]. Here, we report the case of a patient with MND who had dyspnea as the initial symptom, with no weakening of the muscles or muscular atrophy, which ruled out disuse-induced deconditioning.
CASE REPORT
In February 2014, a 61-year-old male with no medical history except for hypertension, showed signs of exertional dyspnea. On worsening of his cough and dyspnea, the patient visited the hospital, where he was treated for his symptoms. In December 2014, aggravation of dyspnea, general weakness, and mild dizziness, in his being admitted to an emergency department, at which point carbon dioxide (CO2) narcosis was observed. Arterial blood gas analysis showed a pH of 7.350, a pCO2 of 63.7 mmHg, and a pO2 of 257.9 mmHg. After intubation, he was transferred to the intensive care unit and placed on a ventilator. Tracheostomy was done in January 2015. A chest X-ray and echocardiogram examination showed no evidence of cardiopulmonary problems. Brain magnetic resonance imaging study, nerve conduction study, and needle electromyography (including the lower cervical, thoracic and lumbar paraspinal muscles as well as bilateral upper and lower extremities) also revealed no abnormalities.
After 24 hours on the ventilator, the general condition of the patient improved; his dyspnea and CO2 narcosis were ameliorated. In May 2015, he was transferred to our hospital for additional investigations to identify the cause of his symptoms, as well as for comprehensive rehabilitation including pulmonary rehabilitation. Motor examination did not reveal any significant weakness, nor was there any indication of muscle atrophy. Sensory functions were intact in all extremities. On functional level evaluation, the patient was only able to sit, due to his dependence on the 24-hour mechanical ventilation via tracheostomy tube, and his muscles being in a state of disuse. Initial laboratory study revealed no abnormalities. Arterial blood gas analysis was also within the normal range with use of the ventilator.
Even though physical examination showed nothing except muscle deconditioning, other examinations revealed prominent diaphragm. Pulmonary function test recorded his vital capacity at 420 mL (11.8% of the predicted value) when sitting, and 350 mL (9.8% of the predicted value) while in a supine position, as measured with a tracheostomy tube. His peak cough flow was too low to be checked. When given a diaphragmatic movement evaluation through fluoroscopy, a 0.25 cm diaphragm movement was present on both sides while the patient was at rest, and there was no difference in maximal breathing from resting status. Also, an additional nerve conduction study done on the bilateral phrenic nerves to check phrenic nerve-diaphragm function showed a prolonged latency and low amplitude of compound motor action potentials (Table 1). Needle electromyography on both sides of the diaphragm showed abnormal spontaneous activity and a partial interference pattern on full inspiration. Follow-up nerve conduction studies and needle electromyography revealed a widespread active neurogenic process in the brain stem, and the cervical, thoracic, and lumbar segments of the spinal cord.
Although his initial symptom was simple respiratory muscle weakness, the patient was diagnosed with MND. A genetic study was conducted to ensure an accurate diagnosis. However, nothing unusual was found, such as superoxide dismutase 1 (SOD1) mutation, a spinal and bulbar muscular atrophy (SBMA) triplet repeat, or a survival motor neuron 1 (SMN1) gene mutation.
Although speech evaluation showed a mild degree of dysarthria, his articulation was nearly perfect; he scored a 93.0% in phonation accuracy. Mild penetration without definite laryngeal aspiration, evaluated with small and large liquid, was observed on videofluoroscopic swallowing studies, thus concluding that the patient did not have bulbar muscle dysfunction.
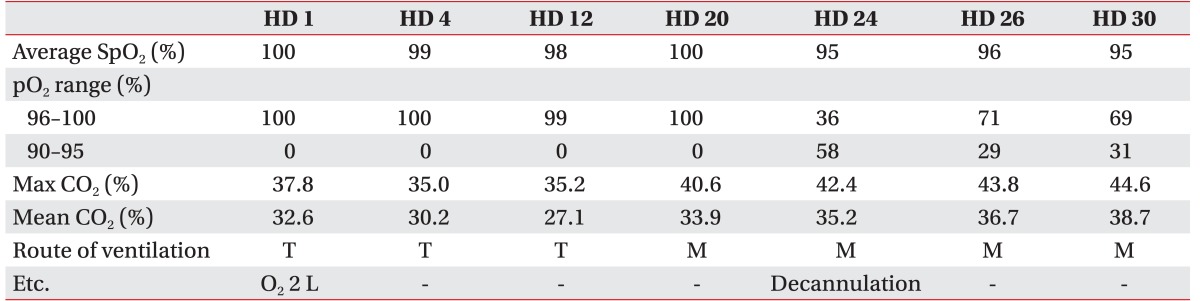
The non-fenestrated cuffed tracheostomy tube was changed to a fenestrated cuffless tracheostomy tube, and noninvasive ventilation training through a nasal mask was conducted. There were no complaints of dyspnea or dizziness, and CO2 monitoring showed no retention (Table 2). Therefore, a decannulation was conducted on April 13, 2015, as the patient was able to tolerate tracheostomy tube capping and noninvasive ventilation. The patient no longer required ventilation during the daytime, but noninvasive ventilation was continued at night to alleviate nocturnal respiratory problems.
Discontinuation of ventilator during daytime, along with comprehensive pulmonary rehabilitation, contributed to recovery in general and respiratory functions. Upon discharge, the patient had recovered extensively, performing nearly independent activities of daily living such as hand writing, using chopsticks and walking with minimal assistance. A follow-up fluoroscopy revealed a slight improvement, as the patient's diaphragm movement was about 1 cm on both sides at rest, and 1.5 cm at both sides on maximal breathing; however, the diaphragm weakness persisted and continued to cause nocturnal hypercapnia, and hence respiratory assistance was continued by noninvasive ventilation at night.
DISCUSSION
Although dyspnea is a result of severe respiratory muscle weakness, and is an advanced-stage complication of MNDs and major cause of fatality, it is seldom an initial symptom. Very rarely does a patient require mechanical ventilation due to acute respiratory failure before being diagnosed with MND. Respiratory failure for amyotrophic lateral sclerosis (ALS), the most common type of MND, is also a late complication. However, with adequate assistance for breathing using noninvasive ventilation, patients with respiratory onset ALS may not show a rapid progressive course [7]. Broadly, this could be similar in MNDs with respiratory onset.
Due to bilateral phrenic failure, most patients present with dyspnea, orthopnea, and hypercapnia [3456], and it is extremely uncommon for the neurological examination to reveal any other conditions [68]. There have been no reports in Korean patients presenting with dyspnea in the absence of other characteristic symptoms of MND, such as limb weakness, muscle atrophy, dysarthria, or dysphagia.
Most physicians will not consider a diagnosis of MND until its characteristic clinical features are evident. Many patients experience respiratory failure in conjunction with increased muscle tension, muscle weakness, muscular atrophy, and fasciculation, and cases involving only acute respiratory failure are extremely rare [8]. Our patient had no muscular atrophy or bulbar dysfunction, and the results of an initial neuromuscular conductivity test were normal; he only had a prior history of requiring a ventilator to treat dyspnea, orthopnea, and hypercapnia, making it difficult to diagnose MND.
In a previous case, a Korean patient diagnosed with MND was applied to mechanical ventilation through a tracheostomy site [9]. However, there are many disadvantages of tracheostomy, including limitation in swallowing and conversation, and consequently a low quality of life. Therefore, if necessary, mechanical ventilation by noninvasive method should be considered.
In conclusion, in the absence of limb or bulbar involvement, when diaphragmatic weakness appears as the initial symptom in patients with acute respiratory failure of unknown cause, MNDs should be considered and be investigated with a neuromuscular conductivity test that includes assessment of the phrenic nerve. Patients thus diagnosed will exhibit notable improvement after being given appropriate respiratory assistance.

 XML Download
XML Download