

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a disease that progresses rapidly after symptom development in most cases, causing death within 2–3 years [1]. In addition to rapid disease progression, its early diagnosis is difficult, and there are only a few treatments that are effective in extending the lifespan of patients. To date, the 5-year survival rate and median survival after diagnosis reported in the literature are approximately 28% and 2.5 years, respectively, and it is <1 year in patients with bulbar symptoms [2345]. Recent studies have reported an improved survival rate [678], presumably as a result of treatments such as the use of mechanical ventilation, tracheostomy, percutaneous gastrostomy, and riluzole. A study by Pupillo et al. [7] reported discordant results regarding ALS survival rate, probably because of the number of samples, certainty of diagnosis, criteria for diagnosis, and differences among the follow-up periods. Furthermore, to determine the efficacy of recent attempts made using new approaches such as diaphragmatic pacing [910], stem-cell therapy [11], and growth factor induction [12], accurate information on the actual outcome and survival should be provided beforehand.
The institute in which this study was undertaken is the first pulmonary rehabilitation center in Korea started in 2001. Majority of the domestic patients diagnosed with ALS visited the institute for consultation on pulmonary rehabilitation, and the ventilator was applied in a timely manner as a non-invasive method in patients who had missed the appropriate time of starting treatment, which may happen often because of their aversion to invasive treatments resulting in a long-term extension of the lifespan. The patients were also approached in a multidisciplinary way by co-work of the physician, physical therapist, occupational therapist, psychotherapist, speech therapist, and social worker during each admission period. Therefore, this study aimed to analyze the survival rate for comparison with previous results and to assess the factors that affect survival under the assumption that the survival rate of ALS patients who were followed up while receiving a relatively consistent standard of treatment at this single institute after diagnosis would show improved outcomes compared with the outcomes shown in other previously published studies.
MATERIALS AND METHODS
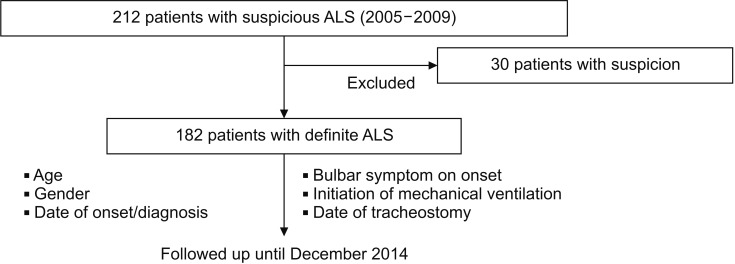
A retrospective analysis of the medical records at this single institute was performed for patients with suspected ALS who visited the institute between January 2005 and December 2009 to examine the survival rate and the factors that affect survival. After reviewing their medical records, only those who were diagnosed based on the revised El Escorial criteria [13] as having clinically definite ALS, clinically probable ALS, and clinically probable along with laboratory supported ALS were included (Appendix 1), whereas those with a doubtful diagnosis who were strongly suspected to have other conditions were excluded from the follow-up. This study was conducted under the approval of the Institutional Review Board of Gangnam Severance Hospital (No. 3-2015-0192).
Using the admission summary and outpatient medical records of patients, the following parameters were examined: time of symptom (such as weakness, fasciculation, dysarthria, or dysphagia) onset, time of diagnosis, bulbar invasion at the time of diagnosis, application of the ventilator and time of the application, the way in which ventilator was used, application of tracheostomy or percutaneous gastrostomy, and survival. The medical records up to December 2014 were used to determine whether the patients were followed up; for those who were lost to follow-up at this pulmonary rehabilitation center, the usage data provided by ventilator rental companies as well as the data provided by homecare nurses based on their home visits were used to confirm death, survival, and continued use of ventilators. Fig. 1 shows the procedure of this study.
Statistical Package for the Social Science (SPSS) ver. 20.0 software (IBM, Armonk, NY, USA) was used for statistical analysis. The survival rate was analyzed using the Kaplan-Meier survival curve, whereas the effect of each factor on the survival rate and prognosis was analyzed using the Cox proportional hazards model. The statistical significance level was set at p<0.05.
RESULTS
General characteristics of patients
The present study included 212 patients with suspected ALS who visited this single pulmonary rehabilitation center between January 2005 and December 2009. After patients with other diagnoses were excluded, the final number of patients confirmed based on the diagnostic criteria was 182. These 182 patients were followed up until December 2014 (Fig. 1).
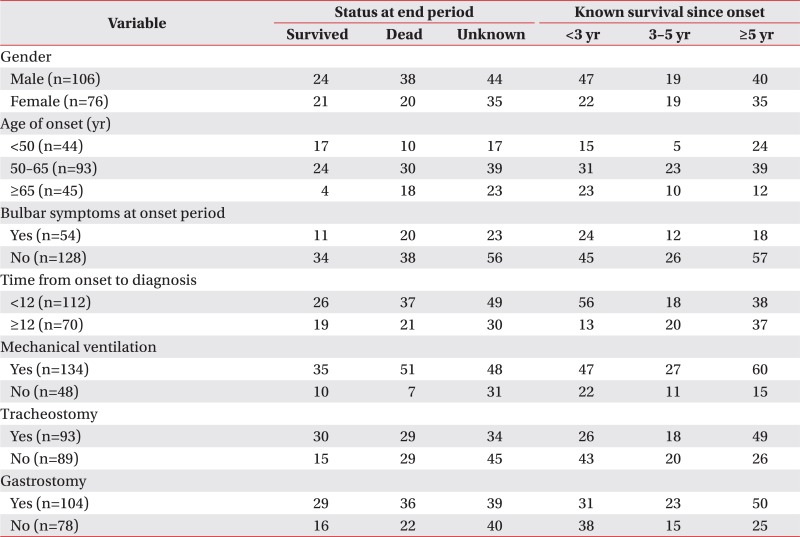
Patients included 106 males and 76 females. At the time of onset, 44 patients were aged below 50 years, 93 patients were aged between 50 and 65 years, and 45 patients were aged 65 years or older. At the time of symptom onset, 54 patients had accompanying bulbar symptoms. Among these 182 patients, 134 received the application of a mechanical ventilator; among these 134 patients, 90 received tracheostomy. In addition, 3 patients who did not receive the application of a mechanical ventilator received tracheostomy. Further, 104 patients underwent gastrostomy. The general characteristics of patients are summarized in Table 1 and additional information is listed separately in Appendix 2.
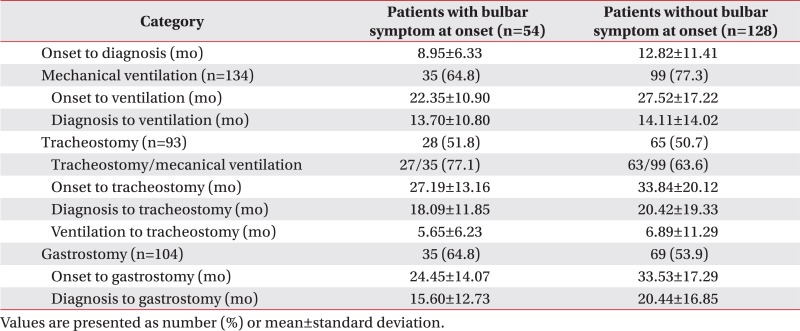
On comparing the patients who had accompanying bulbar symptoms at onset (n=54) with those who did not have accompanying bulbar symptoms (n=128), patients who had bulbar symptoms tended to be started on mechanical ventilation earlier (22.35±10.90 months vs. 27.52±17.22 months from onset), receive tracheostomy earlier (27.19±13.16 months vs. 33.84±20.12 months from onset), and receive gastrostomy earlier (24.45±14.07 months vs. 33.53±17.29 months from onset). Among the 54 patients with accompanying bulbar symptoms at onset, 35 patients were applied to a mechanical ventilator; whereas 99 out of the 128 patients without accompanying bulbar symptoms were applied to a mechanical ventilator. However, among the 35 patients with bulbar symptoms at the onset period who were applied to a mechanical ventilator, nearly 80% of them underwent tracheostomy (27/35, 77.1%), while 60% of the patients underwent tracheostomy (63/99, 63.6%) among those without bulbar symptoms at the onset period. The additional information is listed in Appendix 3.
Analysis of the survival rate
Patients were diagnosed at an average time period of 12.0 months since symptom onset and they had an average subsequent follow-up period of 52.5 months. The number of patients who were diagnosed within 12 months of symptom onset was 112, and the number of patients who were diagnosed 12 months after the symptom onset was 70. A total of 58 patients died during the follow-up period, survival until December 2014 was confirmed in 45 patients, and 79 patients were lost to follow-up. A total of 75 patients could be followed up for at least 5 years since the disease onset, and 38 patients were followed up for at least 3 years since the disease onset but they died or were lost to follow-up within 5 years; the remaining 69 patients died or were lost to follow-up within 3 years since the disease onset.
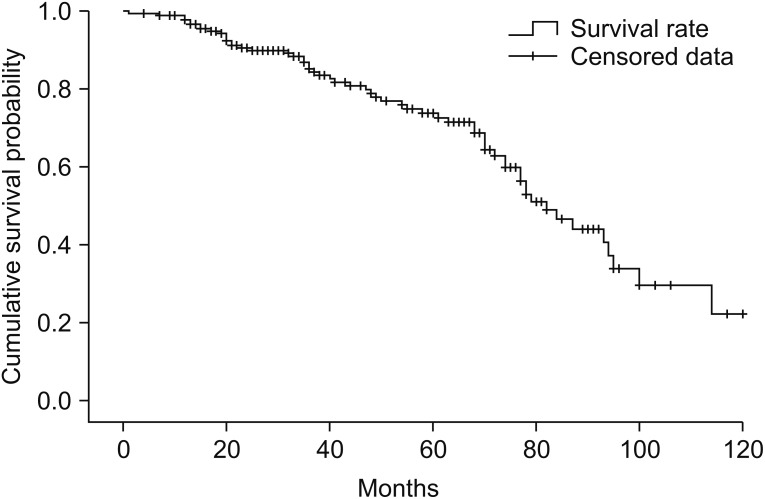
The mean 3- and 5-year cumulative survival rates of patients after symptom onset were 61.5% and 40.1%, respectively; after diagnosis, the mean 3- and 5-year cumulative survival rates were 49.5% and 24.2%, respectively. Among the 182 patients, the mean survival time was 85.42±3.50 months after symptom onset and 75.22±3.76 months after diagnosis.
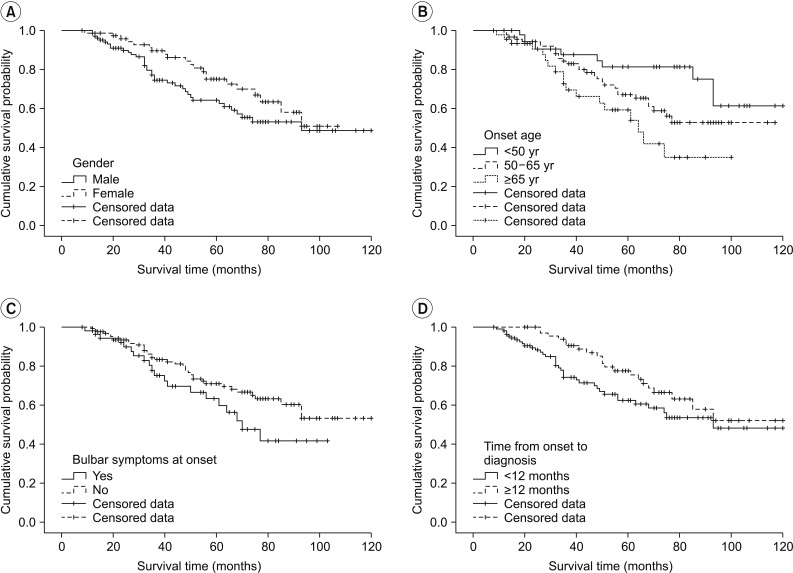
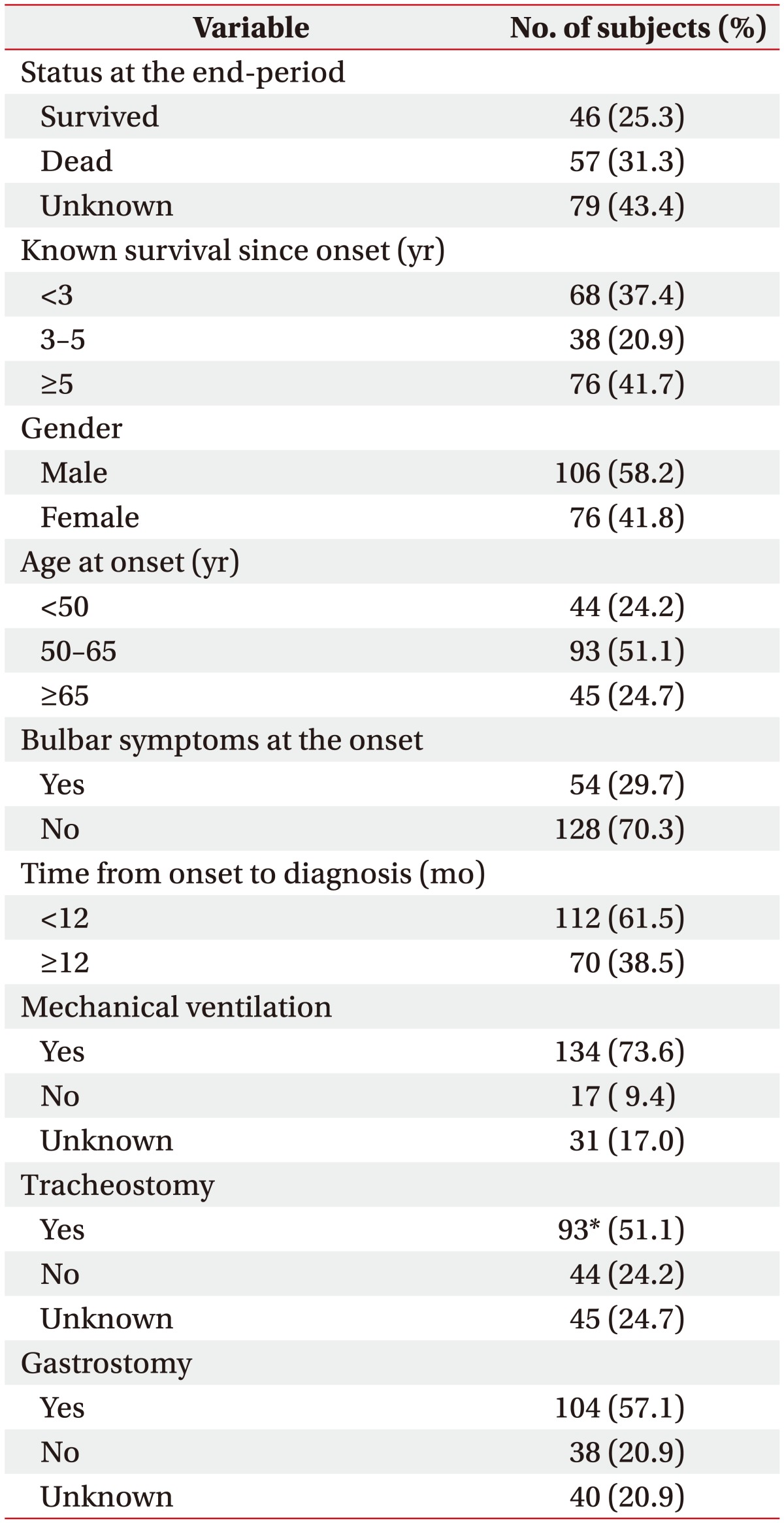
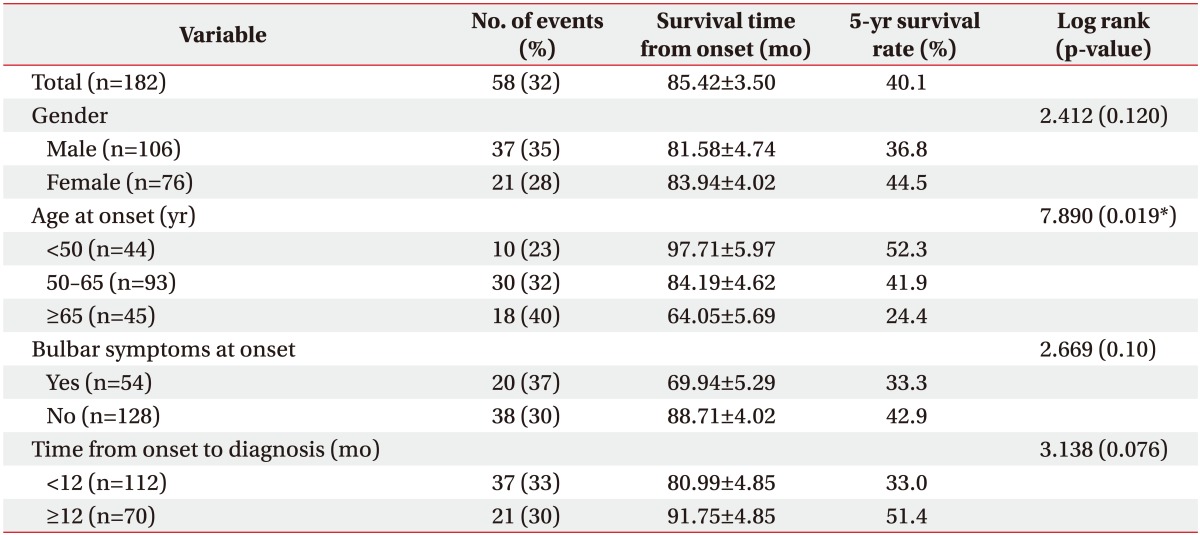
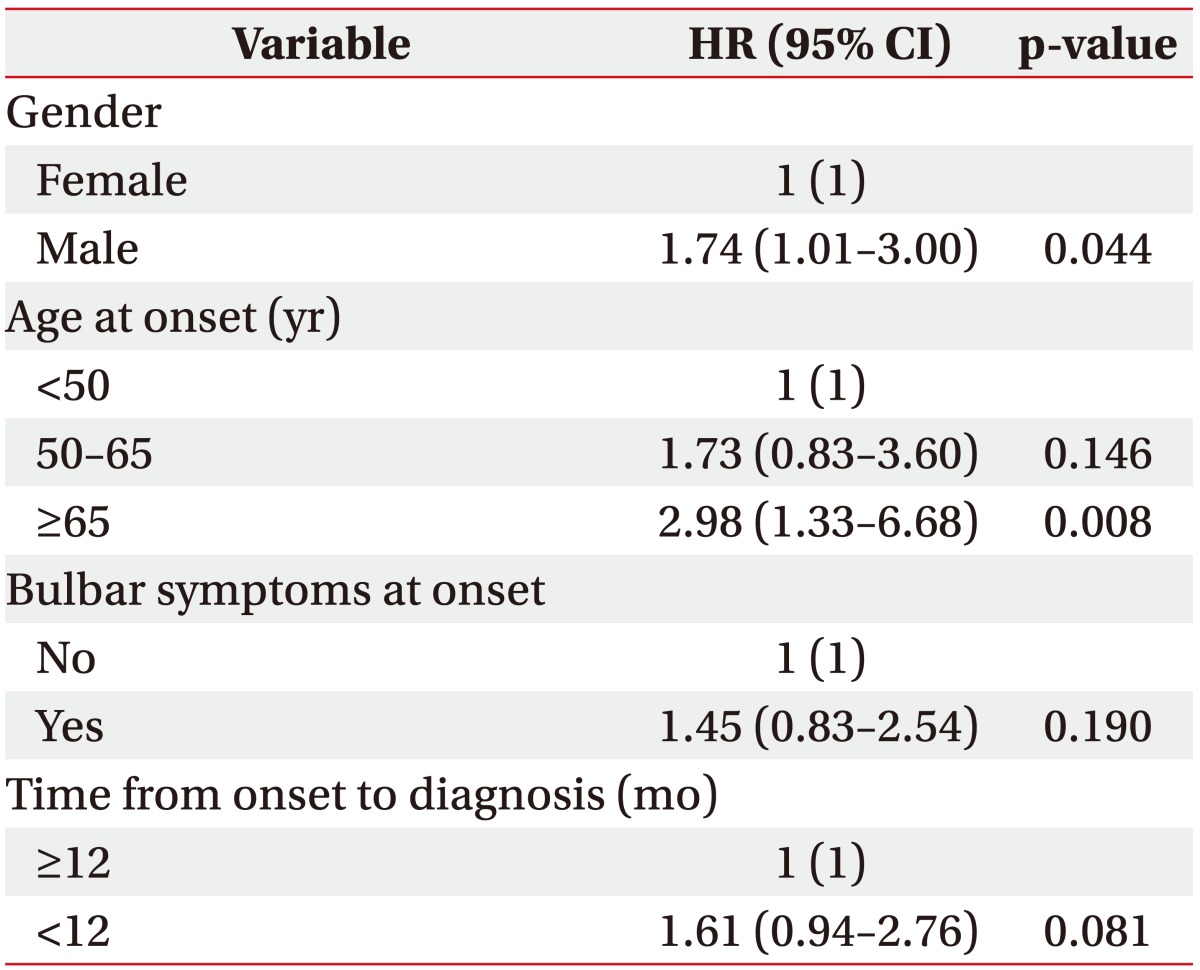
When examined based on sex, the mean survival time was longer in females, with a survival time of 81.58±4.74 months in males and 83.94±4.02 months in females; in addition, the 5-year cumulative survival rate was higher in females (44.5%) compared with males (36.8%) (log rank=2.412, p=0.120). In terms of the age at onset, the mean survival time was the longest in those under the age of 50 years at the time of disease onset (97.71±5.97 months, 5-year cumulative survival rate 52.3%); in those who were aged between 50 and 65 years at onset, the mean survival time was 84.19±4.62 months (5-year cumulative survival rate 41.9%), which significantly dropped to 64.05±5.69 months in those aged above 65 years at onset (5-year cumulative survival rate 24.4%; log rank=7.890, p=0.019). With respect to bulbar symptoms, the mean survival time was 69.94±5.29 months (5-year cumulative survival rate 33.3%) in patients with bulbar muscle paralysis; the survival time was longer in those without bulbar muscle paralysis (88.71±4.02 months, 5-year cumulative survival rate 42.9%; log rank=2.669, p=0.10). In terms of the time from disease onset to diagnosis, the mean survival time was 80.99±4.85 months (5-year cumulative survival rate 33.0%) in those diagnosed within 12 months of symptom onset, and the mean survival time was 91.75±4.85 months in those diagnosed after 12 months from onset (5-year cumulative survival rate 51.4%; log rank=3.138, p=0.076). The cumulative survival rate of all patients is shown in Fig. 2 using the Kaplan-Meier analysis, and the differences in the cumulative survival rate depending on various factors are presented in Fig. 3. The results of analyzing the mean survival time and the cumulative 5-year survival rate are summarized in Table 2.
Analysis of prognosis
Analysis using the Cox proportional hazards model showed that the risk of death after onset was significantly higher in males than in females (hazard ratio [HR]=1.74; 95% confidence interval [CI], 1.01–3.00; p=0.044). The risk also increased with increasing age at onset, being higher in those between 50 and 65 years of age at onset than in those below the age of 50 years at onset (HR=1.73; 95% CI, 0.83–3.60; p=0.146), and the risk was significantly higher in those above 65 years of age at onset than in those below 50 years of age at onset (HR=2.98; 95% CI, 1.33–6.68; p=0.008). On the other hand, cases with bulbar muscle paralysis at onset showed an increased risk compared with cases without bulbar muscle paralysis (HR=1.45; 95% CI, 0.83–2.54; p=0.190); however, the difference was not statistically significant. Furthermore, the risk tended to be higher in those diagnosed within 12 months from disease onset than in those diagnosed after 12 months from onset (HR=1.61; 95% CI, 0.94–2.76; p=0.081); however, the difference was not statistically significant. The details of risk analysis using the Cox proportional hazards model are listed in Table 3.
DISCUSSION
This study analyzed the factors that affect the survival and survival rate by retrospectively following up ALS patients who visited this institute between January 2005 and December 2009. As mentioned earlier, several studies have been conducted to assess the survival rate of ALS patients [134678141516]. However, long-term follow-up studies assessing the survival rate in a large number of ALS patients have not been published in South Korea [7], and no studies have been published recently since various conservative treatments, including drug therapy, have been introduced. The significance of this study lies in the fact that this is the first study to be conducted since the 2000s to assess the survival rate in domestic ALS patients.
In the present study, the mean survival time after symptom onset was 85.42±3.50 months and the mean survival time after diagnosis was 75.22±3.76 months. The 5-year survival rate after symptom onset was 40.1% and the 5-year survival rate after diagnosis was 24.2%. The 5-year survival rate in patients reported in the 1990s was approximately 28% [234], and the percentage still seems to be approximately 22%–28% since symptom onset in studies published since the 2000s [7], excluding the study conducted by Lee et al. [14]. The study by Lee et al. [14] reported that the mean survival time and the 5-year cumulative survival rate in 1,140 ALS patients from Taiwan were 67 months and 52%, respectively; the authors of the above study explained that the difference in the results was because of the use of riluzole and tracheostomy. Furthermore, the mean age of patients at diagnosis was relatively low (56 years), and the fact that only 17% of patients were over the age of 70 years would have influenced the results. Keeping the study by Lee et al. [14] aside, this study shows an improved survival rate compared with the survival rates reported in numerous studies conducted previously. This is believed to be the result of our multidisciplinary approach toward ALS patients, the use of riluzole and adjuvant therapies, including the ventilator, nutritional supply, and percutaneous gastrostomy. The use of riluzole in patients could not be analyzed owing to insufficient records. However, besides this, treatment based on the multidisciplinary approach was already in place from the start of the study in 2005 with the help of not only treating physicians and nurses but also physical therapists, psychotherapists, occupational therapists, and speech therapists. In addition, the ventilator has been actively used to help patients with breathing since the 2000s; 134 of the 182 patients (73.6%) received respiratory assistance with the application of a mechanical ventilator after an average of approximately 2.2 years since the disease onset and approximately 1.2 years since the diagnosis. Among the 75 patients who have survived more than 5 years, 60 patients were with mechanical ventilation. Moreover, 104 patients (57.1%) who received percutaneous gastrostomy for tube feeding would have had a considerable impact on improving the survival rate. In 2011, Spataro et al. [17] reported that percutaneous gastrostomy improves survival in dysphagic ALS patients. Also, a recent study by Fasano et al. [18] reported a gain of tracheostomy-free survival in patients who underwent percutaneous gastrostomy, and it also reported a greater survival if the percutaneous gastrostomy tube is inserted before a significant weight loss occurs.
Upon analyzing the factors affecting survival, the present study showed higher age as the predictor of poor prognosis, which is consistent with the results observed in other studies [781416]. Also, this study showed male gender as the predictor of poor prognosis. In previous reports, some authors have reported better survival in men [719], and some authors have reported worse survival in men although the difference was not statistically significant [81416]. In this study, however, men showed worse prognosis for survival, even with a smaller proportion of those aged over 65 years compared to women (21.6% in men vs. 28.9% in women).
Although the risk tended to increase in patients who had bulbar muscle paralysis at onset or were diagnosed within 12 months of onset, the difference was not statistically significant. Previously published studies have shown that the survival rate decreased significantly when bulbar muscle paralysis was present than when bulbar muscle paralysis was absent [716]. We believe that the reasons for not observing a significant decrease in the survival rate despite bulbar muscle paralysis in this study were adequate airway secretion management and breathing assistance through tracheostomy and nutrition supply through percutaneous gastrostomy. In general, patients with bulbar muscle paralysis frequently experience swallowing disorders, leading to difficulty in oral intake of nutrition, and although some patients maintain ventilators for long term in a non-invasive manner, many of them encounter limitations. For these reasons, the prognosis is generally worse when bulbar muscle paralysis is present. Based on the analysis of results obtained from this study, in comparison with the application of ventilators, tracheostomy was more frequently performed in patients who initially exhibited bulbar muscle paralysis, and the frequency of the percutaneous gastrostomy procedure was also higher in these patients when compared with those who did not exhibit bulbar muscle paralysis. Also, tracheostomy (27.2 months vs. 33.8 months since symptom onset) and gastrostomy (24.5 months vs. 33.5 months since symptom onset) were performed much earlier in patients with initial bulbar muscle paralysis than in patients without initial bulbar muscle paralysis. Thus, it can be deduced that these appropriate treatments resulted in a survival rate that did not show a big difference between those with and without bulbar muscle paralysis despite the known worse prognosis in those with this condition. In patients who received an early diagnosis, better prognosis was predicted compared with that in those who did not receive an early diagnosis; however, the survival rate was even lower, which may have resulted from rapid progression of symptoms leading to a relatively evident diagnosis.
The limitation of the study was limited access to patients who were lost to follow-up because this was a retrospective study. On study completion in December 2014, 79 out of 182 patients were lost to long-term follow-up and their survival could not be determined, resulting in cropped data. However, the Kaplan-Meier analysis that included those who were lost to follow-up based on the last data point at which survival could be determined; even when all of them were assumed to be dead after their last visit, the 5-year survival rate of 40.1% after symptom onset and the 5-year survival rate of 24.2% after diagnosis would not change. These values, as mentioned previously, inarguably show impressive improvement compared to previous results. Moreover, results of this study did not consider the possible risk of concomitant diseases and this point should be considered in future studies.
Although being a retrospective study, this study is the first study to include a long-term follow-up for survival in a large number of domestic ALS patients since the 2000s. For ALS patients who visited our institute, a marked improvement in survival was observed following the recent use of mechanical ventilator, riluzole, and adequate nutrition. Although prognosis for survival is poor in males with a higher age at onset, patients with bulbar symptoms at onset did not show a big difference in the survival rate from those without these symptoms. Although the prognosis of ALS patients is generally known to be poor, the survival rate of patients in this study was found to be excellent compared with the survival rate of patients in previous studies.

 XML Download
XML Download