

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Guillain-Barre syndrome (GBS) is a condition characterized by an acute or subacute onset of varying degrees of weakness in the limbs or cranial nerve-innervated muscles, associated with decreased or absent deep tendon reflexes, and a characteristic profile in the cerebrospinal fluid and electrophysiologic studies [1]. Advances in general care facilities and the availability of specific treatments have improved the outcome for patients with GBS. Nonetheless, approximately 20% of patients die from complications associated with GBS or remain severely disabled as a result of this condition [23].
The concept of GBS changed substantially in the 1990s because an axonal subtype of this disorder, acute motor axonal neuropathy (AMAN), was identified in northern China [4]. Consequently, GBS could be divided into two major subtypes, acute inflammatory demyelinating polyneuropathy (AIDP) and AMAN [56]. The pattern and speed of progression differ between AMAN and AIDP. Significantly shorter mean periods from onset to peak of illness are seen in AMAN [7]. Traditionally, AMAN has been thought to have a poorer clinical outcome [89]. However, some recent reports have shown that patients with AMAN can also achieve rapid recovery. In contrast to the relatively uniform speed of recovery in patients with AIDP, two different patterns of recovery are seen in patients with AMAN. Some recover within months; whereas, others have a slow and poor recovery [101112]. Rapid recovery is caused by resolution of the conduction block, and poor recovery is associated with extensive axonal degeneration at the nerve roots [13].
Previous studies showed that preceding infection, age, rapid progression, disability at the nadir, disability at 2 weeks after entry, and electrophysiological characteristics are associated with long-term prognosis [3141516171819]. However, most of the studies assessing the prognostic factors in GBS were performed mainly in Europe and North America, where a diagnosis of AMAN is made in only 3%–17% of cases of GBS whereas the AIDP subtype accounts for 69%–90% of patients [202122]. Notably, higher proportions of patients with GBS in East Asia and South America are classified into the axonal type [2324]. This geographic difference in the proportion of the two disease subtypes suggests that prior studies that assessed the prognostic factors of GBS were based primarily on AIDP patients.
Unlike AIDP, in the AMAN subtype of GBS, autoimmune attack occurs at the nodes first and then extends to the paranodes [25]. The immunologic target of axonal and demyelinating GBS is different; hence, the prognostic factors may differ between these two types of GBS. However, virtually no research has been conducted to assess the factors associated with rapid recovery in patients with the axonal type of GBS. The aim of our current study was to identify the factors that can predict the functional outcomes for patients with axonal GBS.
MATERIALS AND METHODS
Study design
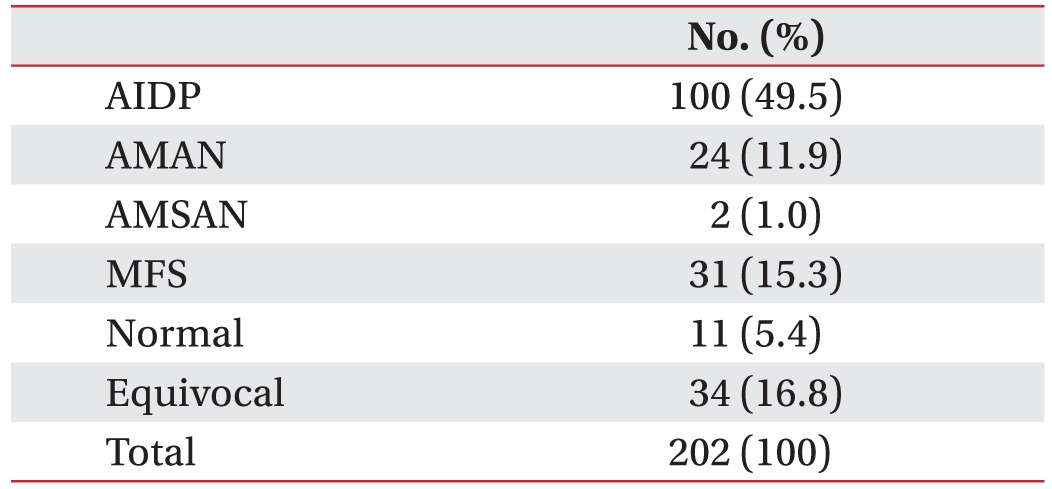
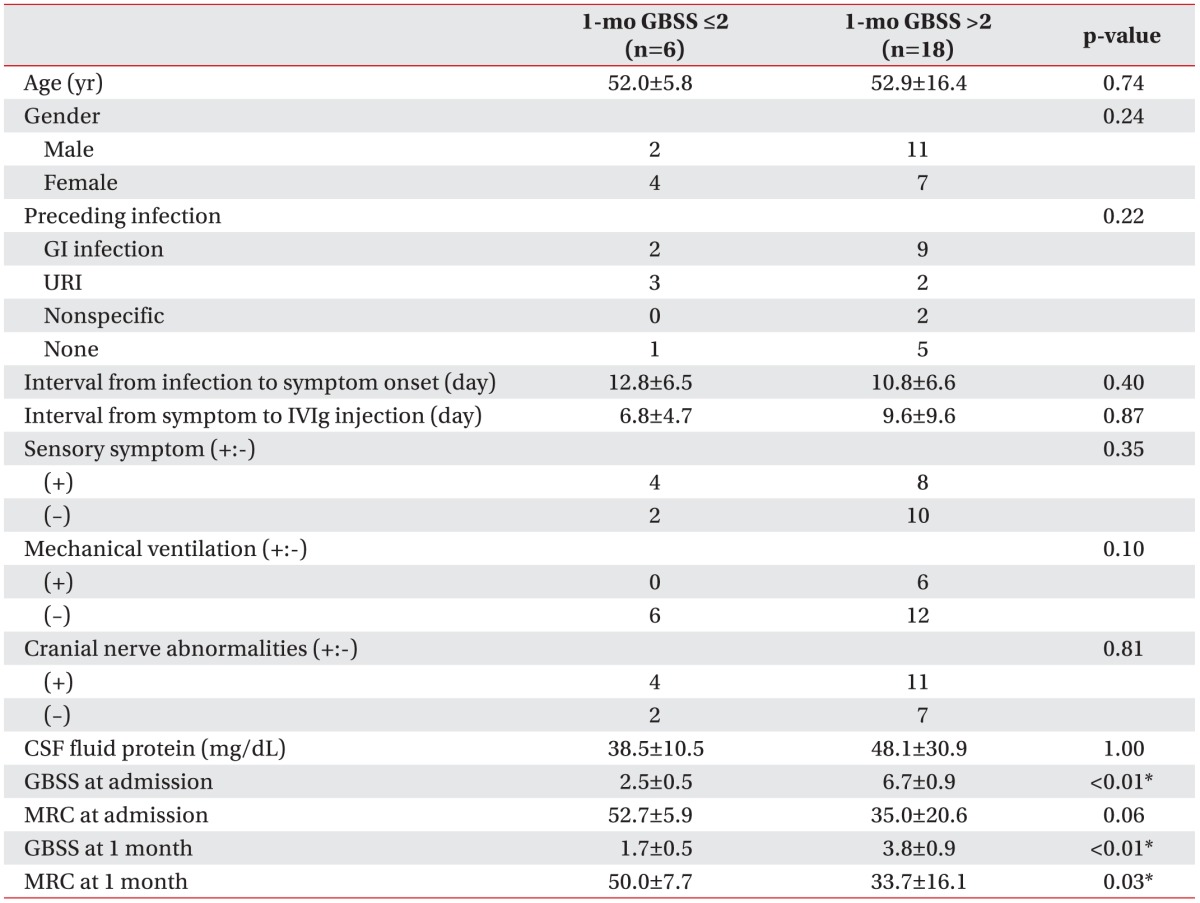
Two hundred and two patients admitted to our university hospital between January 2003 and December 2014 were retrospectively reviewed according to the following criteria: (1) above 18 years of age; (2) first occurrence of GBS; (3) fulfilled the National Institute of Neurological Disorders and Stroke diagnostic criteria for GBS [26]; (4) fulfilled the Van den Bergh criteria for the axonal type of GBS [27]; and (5) classified as having acute motor axonal neuropathy. Patients who were previously diagnosed with other peripheral neuropathies were excluded. We defined a poor outcome as the inability to walk independently at 1 month after admission (GBS disability score ≥3) and a good outcome as being able to walk independently at 1 month after admission (GBS disability score ≤2). These measures have been used as primary endpoints in many other previous studies on GBS [3282930].
Classification of axonal GBS according to the van den Bergh criteria
In the Van den Bergh criteria, the diagnosis of AIDP is supported by >70% degree of slowing of motor conduction velocity below the lower limit of normal values, >150% prolongation of motor distal latency above the upper limit of normal values, >120% prolongation of the F latency above the upper limit of its normal value (and >150% prolongation, if the distal negative peak compound muscle action potential [CMAP] amplitude was <80% of the lower limit of normal values), or abnormal temporal dispersion (i.e., >30% negative peak CMAP duration increase) in two or more nerves. For the axonal form of GBS, there should be no features of AIDP and distal CMAP should be <80% of the lower limit of its normal value in two nerves.
Data collection
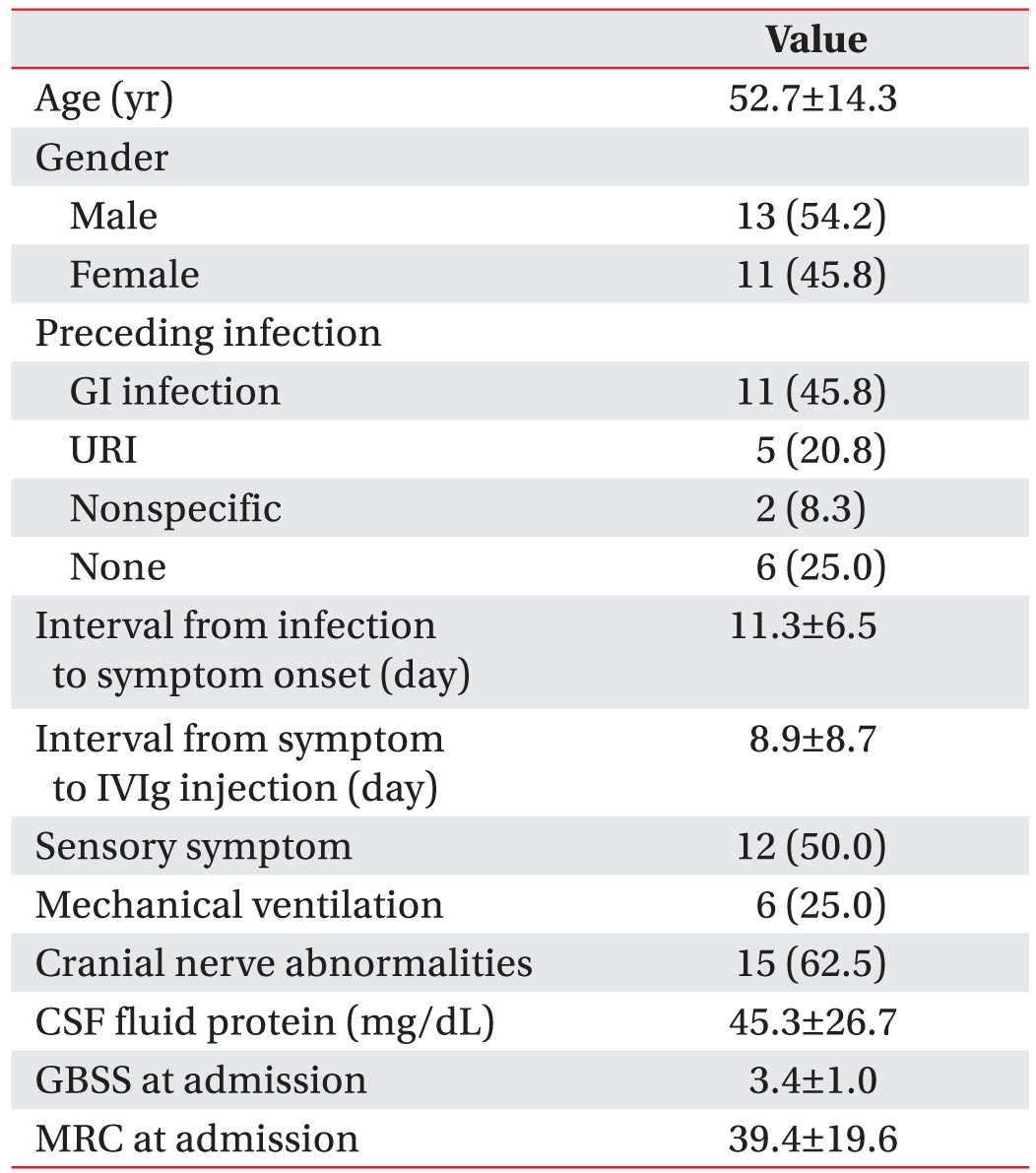
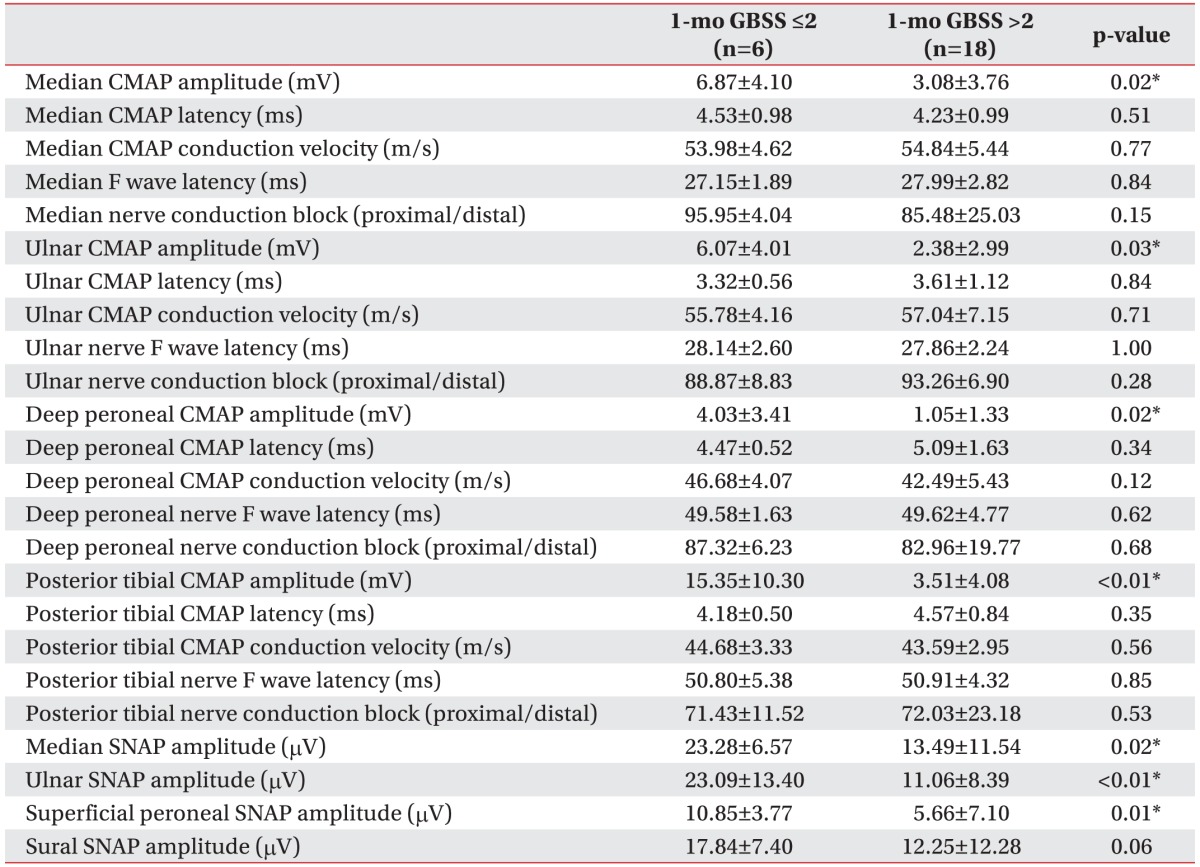
Data regarding patient age, gender, type of preceding infection, interval from infection to symptom onset, interval from symptom onset to intravenous gamma-globulin (IVIg) injection, cranial nerve dysfunction, sensory disturbance, cerebrospinal fluid protein, and respiratory disturbance requiring mechanical ventilation were reviewed. In addition, Medical Research Council (MRC) sum scores and GBS disability scores at admission and 1 month after admission were documented. The MRC sum score is defined as the sum of MRC scores of six different muscles measured bilaterally, which results in a sum score ranging from 0 (quadriplegic) to 60 (normal) [31]. The GBS disability score is a functional scale for patients with GBS, ranging from 0 (normal) to 6 (death) [32]. Two points in the GBS disability score indicates an ability to walk more than 10 m without assistance but an inability to run. Electrophysiologic data involving the amplitude of CMAP; conduction velocity; distal motor latency; minimal F wave latency; conduction block of the median, ulnar, deep peroneal, and posterior tibial nerves; and the amplitude of sensory nerve action potential (SNAP) of the median, ulnar, superficial peroneal, and sural nerves was included. Abnormal nerve conduction study measures were defined as those with amplitudes lower than, and slowed nerve conduction velocity relative to the standard values of our electrodiagnostic laboratory. Motor conduction block was defined as >30% negative peak amplitude reduction of proximal CMAP [27] and the value was recorded as the ratio of the proximal to the distal CMAP amplitude. When a patient had more than one follow-up electrodiagnostic study, the recordings that were considered the most informative were utilized for the final electrodiagnosis and evaluation.
Statistical analysis
Data were analyzed using the Statistical Package for Social Sciences software package (SPSS ver. 18.0; SPSS Inc., Chicago, IL, USA). The Mann-Whitney U test and chi-square test were used to analyze demographic, clinical, and electrophysiologic differences in the good and poor outcome groups. Statistical significance was determined at p-values <0.05.
RESULTS
We enrolled 202 GBS patients between 2003 and 2014. Twenty-six patients (13%) were categorized into the axonal type (Table 1). Twenty-four patients were classified into the AMAN type, and two patients were classified into the AMSAN (acute motor sensory axonal neuropathy) type of GBS. From the baseline characteristics of the motor axonal GBS patients, the mean age at onset was 52.7 years. Twenty-five percent of these patients required mechanical ventilation. The mean GBS disability score at admission was 3.4, and the mean MRC sum score at admission was 39.4 (Table 2). At one month after admission, six patients (25%) were able to walk for 10 m without assistance. Ten patients (42%) needed assistance to walk, two patients (8%) were bedridden, and six patients (25%) needed respiratory support. After dividing the patients by their functional outcomes at 1 month, we evaluated the factors that differed between the good and poor outcome groups (Table 3). Significant differences were found in terms of GBS disability score at admission (p<0.01), GBS disability score at 1 month (p<0.01), and the MRC sum score at 1 month (p=0.03). However, mechanical ventilation did not show a significant difference between the two groups (p=0.10).
An electrophysiologic assessment was performed at a mean time period of 6 days after the admission date. In this analysis, the good outcome group showed a greater amplitude of median (p=0.02), ulnar (p=0.03), deep peroneal (p=0.02), and posterior tibial nerve CMAPs (p<0.01) compared with the poor outcome group. In the sensory nerve conduction study, the good outcome group showed a higher amplitude of median (p=0.02), ulnar (p<0.01), superficial peroneal SNAPs (p=0.01) than the poor outcome group (Table 4).
DISCUSSION
In the present study, 75% of the AMAN patients were
unable to walk independently at 1 month after admission.
This is a larger number than that in a previous study
of all GBS patients, which reported that 55% of these cases
could not walk independently at 4 weeks after inclusion
[29]. In the present study, a poor outcome showed a
strong association with the GBS disability score at admission;
amplitude of the CMAP of the median, ulnar, deep
peroneal, and posterior tibial nerves; and amplitude of
the SNAP of the median, ulnar, and superficial peroneal
nerves. This indicates that the disease severity at admission
and the extent of axonal injury expressed as a low amplitude in an electrophysiologic test may be predictive
of the prognosis at 1 month after admission.
There has been some debate as to whether preceding diarrhea is a predictor of poor outcome in GBS. This finding was reported in five studies [315171829] but not in two other reports [1416]. In the present study, the presence of diarrhea before the onset of weakness was not statistically different between the two groups. This finding does not correspond with the well-established concept that Campylobacter jejuni-induced diarrhea and the axonal type of GBS infection are closely associated with each other. Our result can be explained by the fact that not only C. jejuni, but also other non-Campylobacter bacteria or viral infections could represent a common etiological basis of infectious diarrhea and GBS development. Even if C. jejuni-induced diarrhea was a negative prognostic factor, we could not determine whether diarrhea was associated with C. jejuni because we did not have the stool evaluation results. We could not distinguish C. jejuni-induced diarrhea from other nonspecific diarrheas. Non-specific diarrhea did not show any correlation with a poor prognosis in our results.
In contrast to other studies that included other types of GBS patients, old age was not found to be a prognostic factor in the present study. Two previous studies have reported that younger people are more likely to acquire a C. jejuni infection and develop the axonal type of GBS. Also, there has been an epidemic of axonal GBS among children in northern China [4], and a previous study has reported that young men living in rural areas in Bangladesh are mainly affected by the AMAN type of GBS [33]. However, this result is not consistent with our present findings. In our current study series, the mean age of GBS onset was 52.7 years, which is higher than that reported in other studies of GBS. Even though our present investigation included only a small number of AMAN patients, the standard deviation of age for axonal GBS is smaller than that in the other study's data. Although the issue of whether younger or older patients are more susceptible to axonal GBS is not yet clear, we speculated that if a narrower range of age groups was susceptible to axonal GBS than to the entire spectrum of GBS, age might not be a negative prognostic factor.
Cornblath et al. [34] described distal CMAP amplitude as the single best predictor of prognosis through electrodiagnostic testing in 210 of 245 GBS patients. Miller et al. [35] stated that most powerful predictor of a poor outcome in 60 severe GBS patients was a reduced mean CMAP amplitude. In other studies, less than 20% or 10% of the lower limit of normal was found to be more consistent with a poor prognosis. This is concordant with our present study, which showed that the amplitude of median, ulnar, deep peroneal, and posterior tibial CMAPs and the amplitude of median, ulnar, and superficial peroneal SNAPs were higher in the good prognosis group. Besides, the mean amplitude of the CMAP in the poor prognosis group was more than 48% of the lower limit of normal, and the mean amplitude of the SNAP was more than 68% of the lower limit of normal, a far greater value than was expected based on previous studies. A methodological problem may have affected the result. In the present study, we used the Mann-Whitney U test to identify the prognostic factors. The Mann-Whitney test uses the mean values for evaluating statistical significance. In axonal GBS patients, the involved nerve is unique to each individual; the mean amplitude of each nerve can become nonspecific when the value for each of the involved nerves closely correlated with the GBS disability score at 1 month is averaged.
Our present study had several limitations. First, we included a small number patients from our university hospital only. Second, our electrophysiologic evaluation periods were heterogeneous among individuals and most of our GBS patients were assessed through only one or two electrophysiologic studies. Because there were an insufficient number of serial evaluations for electrophysiologic studies, the lack of distinction between reversible conduction failure in the axonal type of GBS and demyelinating conduction block might have led to the incorrect classification of AMAN as AIDP. In another aspect, there is a possibility that patients with the severe demyelinating type of GBS were considered to have the axonal type of GBS. Third, the primary outcome in our patient series was not measured at 1 month after symptom onset, but it was measured at 1 month after admission. Fourth, antiganglioside antibody evaluations and microbiological examination results were not available. Because our study was performed retrospectively, there were also some limitations in terms of data availability. Finally, even though there were several statistically significant factors established between the two study groups by the chi-square and Mann-Whitney U tests, we could not identify any statistically significant factor by logistic regression analysis that was predictive of a poor outcome at 1 month.
The present study is the first study that attempted to identify the prognostic factors among adult patients with motor axonal GBS. Patients with a higher GBS disability score at admission and a low amplitude in the electrophysiologic study had an inability to walk independently at 1 month after admission. However, old age and preceding diarrhea did not correlate with a poor outcome in patients with axonal GBS. Electrophysiological evaluations and initial disease severity are thought to be important predictive factors of early recovery in patients with axonal GBS. Our findings are limited by the relatively small number of patients analyzed, and we were unable to provide more informative results because our analysis was performed retrospectively. Further complementary prospective studies that involve a larger number of cases and evaluate long-term outcomes are warranted.
 XML Download
XML Download