

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Many genetic diseases can be diagnosed precisely with the aid of various genetic technologies. Sanger sequencing, developed in 1977, was the first generation of sequencing technology [1]. While genetic mapping had been possible, its utility was limited due to the involved high cost and low throughput. Next-generation sequencing (NGS) is based on the sequencing-by-synthesis approach, and includes whole-genome sequencing, whole-exome sequencing, and targeted exome sequencing using a panel [2]. NGS can be easily applied to a whole exome, which comprises 1% of the human genome [2].
Clinical exome sequencing (CES) has been recently applied for the diagnosis of rare inherited diseases in Korea; it is a NGS service which provides not only sequence variation data but also clinical interpretation, which aids in reaching a final conclusion with regards to genetic diagnosis. Recently, commercialized probe panels, which are composed of enrich genes with known clinical relevance, have been introduced. Among them, TruSight One Sequencing Panel (Illumina Inc., San Diego, CA, USA) targeting about 4,800 genes provides more comprehensive coverage of genes with clinical relevance, as it mainly focuses on genes based on Human Gene Mutation Database, the Online Mendelian Inheritance in Man catalog, GeneTests.org, etc. It is more suitable for clinical purpose since it reduces burden of whole exome sequencing in terms of time and cost as well as data analysis. CES using TruSight One Sequencing Panel, which enriches about 4,800 genes with clinical relevance, could be applied for diagnosis of diseases with genetic heterogeneity and a few cases have been reported [3].
In the present case, CES played a critical role in the diagnosis of hereditary spastic paraplegia (HSP), which showed compound heterozygous mutations in SPG11. This case indicates the usefulness of CES as a diagnostic tool for genetic diseases such as HSP.
The ethical aspects of this case were reviewed by the University of Catholic Institutional Review Board. Informed consent was obtained from the patient and her parents.
Go to : 
CASE REPORT
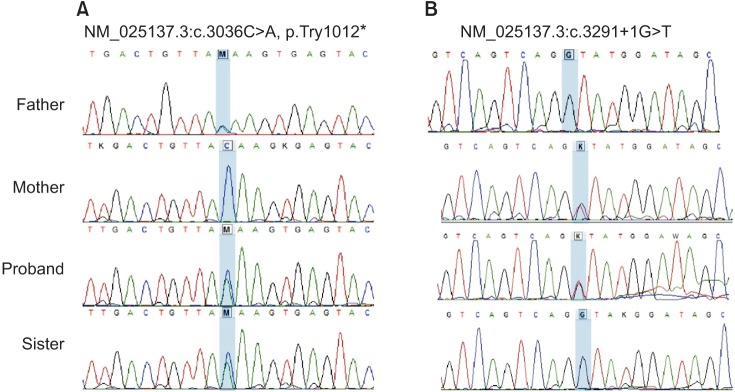
A 17-year-old female visited our physical medicine and rehabilitation clinic complaining of toe walking with cognitive impairment and dysarthria. Her symptoms initiated several years back and progressed slowly; however, she had never had these symptoms evaluated by a medical practitioner. There were no remarkable findings in her genealogical chart; her elder sister had a normal development (Fig. 1), and the patient's birth history was uneventful. On neurological examination, deep tendon reflexes were exaggerated on both the knees and ankles. The Modified Ashworth Scale of knee extension and flexion was grade 2 and 1+, respectively. The motor power of her legs was of fair grade with equinovarus deformities on both the feet, and there was no visible muscle atrophy. Electrodiagnostic studies, including nerve conduction, needle electromyography, and somatosensory and motor evoked potential studies, did not indicate any abnormality. She showed mild cognitive impairment; her Montreal Cognitive Assessment score was 21/30, and her receptive and vocabulary abilities were about 3–6 years lower than those appropriate for her age. The laboratory studies did not reveal any specific findings that could have suggested diseases presenting dysfunction of the central nervous system, such as Wilson's disease, autoimmune diseases, endocrine diseases, or metabolic diseases. Whole spine magnetic resonance imaging (MRI) presented no definite abnormalities; however, brain MRI showed thinning of the corpus callosum (Fig. 2). HSP was suspected based on the clinical manifestations, sporadic spastic paraplegia (SPG), and no evidence of other primary etiology. SPG type 11 was strongly suspected as thinning of the corpus callosum is a common feature in SPG11; however, it is also found in other SPG subtypes. Therefore, sequencing of SPG4 (SPAST) and SPG3A (ATL1), which presents with relatively high frequency among individuals with HSP and SPG11, was performed. Among the 40 coding exons of SPG11, nine exons namely exons 2, 6, 7, 8, 30, 36, 37, 38, and 39 presented relatively high frequency of mutations, and the adjacent introns were analyzed by Sanger sequencing. Sequence analysis of SPG11 in clinical practice is limited in Korea, since safety and effectiveness of the test has not yet been proved by National Evidence-based Healthcare Collaborating Agency. Therefore, genetic evaluation of SPG11 is only available for the purpose of research and not clinical diagnosis. Based on these reasons, we selectively sequenced exons with a high frequency of SPG11, rather than all exons and introns of SPG11 gene. However, gene sequencing and chromosome analyses, including chromosome microarray, did not show any abnormal findings. Thereafter, she visited another clinic to further attempt to find a diagnosis for her disease. A neurologist from another center assumed her disease to be spinocerebellar ataxia (SCA). Fluorescent fragment length analysis of SCA type 1, 2, 3, 6, 7, 8, and 17 was performed; however, no mutations were detected. A few months later, she came to our center again for further detailed genetic studies. Clinically, HSP, especially SPG types 11 and 15, and SCA were suspected. However, DNA sequencing of exons with high incidence mutations did not reveal any matching results. Meanwhile, we were conscious about an NGS technology named CES that can analyze 4,813 genes and about 62,000 exomes associated with many genetic diseases in humans [3]. The CES panel provides comprehensive coverage of HSP and SCA, including 50 of 67 known protein-encoding genes involved in HSP [4] and 33 of 41 genes in SCA although some subtypes of SCA which are a result of trinucleotide repeat expansion could not be accurately detected [5]. Genomic DNA was extracted from the peripheral blood of the patient and her parents and sister. The genomic DNA was enriched using the TruSight One Sequencing Panel, which includes 125,395 probes targeting a 12-Mb region spanning 4,813 genes, and was sequenced on the Illumina MiSeq platform [3]. The size of probe is 80-mer, and it targets libraries of approximately 500 bp enriching 350–650 bases centered symmetrically on the midpoint of the probe. It means that, the panel provides coverage of exon-flanking regions, which is splice sites. Enrichment-ready fragmentation by tagmentation and PCR amplification involved 50 ng of input DNA. Pooled sample libraries were denatured and labeled by biotin-labeled probes specific to the targeted region for hybridization. The streptavidin beads were added and bound to the biotinylated probes. Biotinylated DNA fragments bound to the streptavidin-coated beads were magnetically pulled down from the solution, and the beads were removed. Captured targeted regions were loaded on MiSeq/NextSeq/Hiseq system for sequencing. On-instrument software automatically performed alignment and variant calling. Imported sequence data to the VariantStudio software was filtered and customized reporting for specified genes. CES revealed compound heterozygous variations on the patient's SPG11 gene; a splice site aberration in intron 18 (NM_025137.3:c.3291+1G>T) and a nonsense variation in exon 16 (NM_025137.3:c.3036C>A, p.Try1012*). Each of the variations was reported as a pathogenic variant in the previous reports [67]. The results also included other variants; however, they were not pathogenic but normal variants. Furthermore, Sanger sequencing revealed her parents as being heterozygous carriers for each mutation, and her sister as a carrier of heterozygous mutation (c.3036C>A/p.Try1012*) (Table 1, Fig. 3).
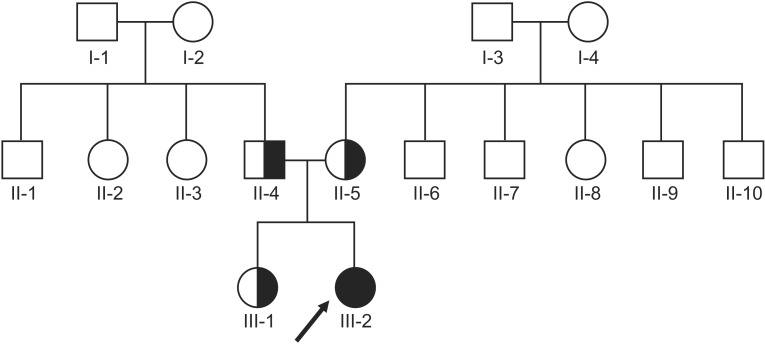 | Fig. 1Pedigree of the family of the 17-year-old female with spastic paraplegia. The black symbol represents the patient. The index I-3 died due to old age, and the index II-6 passed away at an early age due to an unknown cause.
|
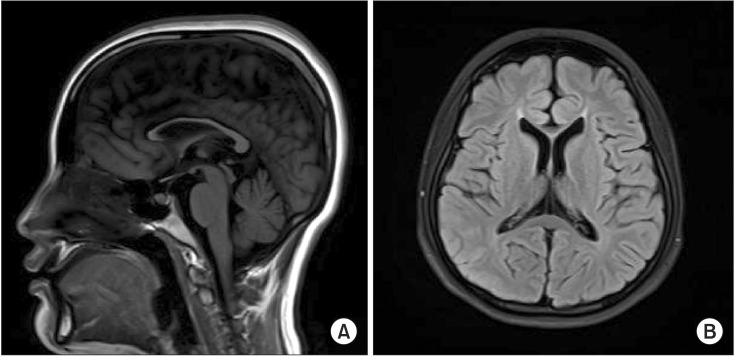 | Fig. 2Brain magnetic resonance imaging findings in the patient. (A) Sagittal T1-weighted image reveals a thin corpus callosum. (B) Axial T2-weighted image also shows a thin corpus callosum.
|

Go to :
DISCUSSION
HSPs are a group of genetic conditions in which spastic paralysis of the legs is the principle clinical feature [8]. They are clinically divided into pure and complicated forms, and genetically divided into autosomal dominant, autosomal recessive, and X-linked recessive forms [9]. At present, genetic mapping and exome sequencing have led to identification of 84 HSP gene loci and 67 known genes causing HSPs [4]. Thinning of the corpus callosum is associated with both SPG11 and SPG15 mutations, and mutations in the former are more common [10]. In our patient, although HSP type 11 was strongly suspected based on clinical features, spastic paraplegia, cognitive deficits and thin corpus callosum, there existed limitation for the evaluation of the SPG11 variants because the mutations in SPG11 are not simply found in a few specific exons; whereas, scattered over 40 exons [10]. Therefore, genetic analysis with whole-exon sequencing of SPG11 using traditional approaches is very time consuming and labor intensive. Moreover, in Korea, genetic analysis of SPG11 has certain limitations imposed by the National Health Insurance Policy; however, few of the variants in SPG11 were analyzed in this report. Before CES was performed in the present case, more money and time was involved in the diagnosis of her disease. CES identified a heterozygous transposition in intron 18 (c.3291+1G>T) and a heterozygous mutation (c.3036C>A/p.Try1012*), which were previously reported as pathogenic [67].
The introduction of next-generation sequencing (NGS) led to an exponential increase in the discovery of genetic causes in both extremely rare and common diseases. Previous cases of HSP have been reported to be diagnosed by whole-exome sequencing or targeted panel sequencing. In Korea, CES with TruSight One Sequencing Panel, which enriches about 4,800 genes with clinical relevance, have been applied for the diagnosis of rare hereditary disease. There is a similar test in the US named as an augmented clinical-exome sequencing test (ACE). The ACE clinical exome test by Personalis Inc. includes more than 8,000 genes related to diseases. The CES and ACE clinical exome tests are designed to provide molecular genetic diagnoses for patients whose clinical assessment indicates a genetic disorder. These tests are appropriate for a variety of exome sequencing indications, providing lower turnaround times, lower cost, and more comprehensive coverage of the target regions without the complexity of whole-exome analysis [3].
In conclusion, it is challenging to diagnose neurologic disorders that involve several genetic mutations. When a specific genetic disorder is suspected, tests are limited to those few mutations with high prevalence. However, if the results are negative, it is difficult to decide on the next step of the diagnosis in terms of whether to proceed to more detailed gene analysis for the same disease or to consider other possible diseases. In this situation, CES can be helpful. In our patient, until CES was introduced, genetic analysis required more time and money (Table 2). The result of CES takes about 2 weeks, and it costs about $1,250 to $1,650. We believe that CES will be a useful tool for the diagnosis of known Mendelian genetic diseases such as HSP and SCA.
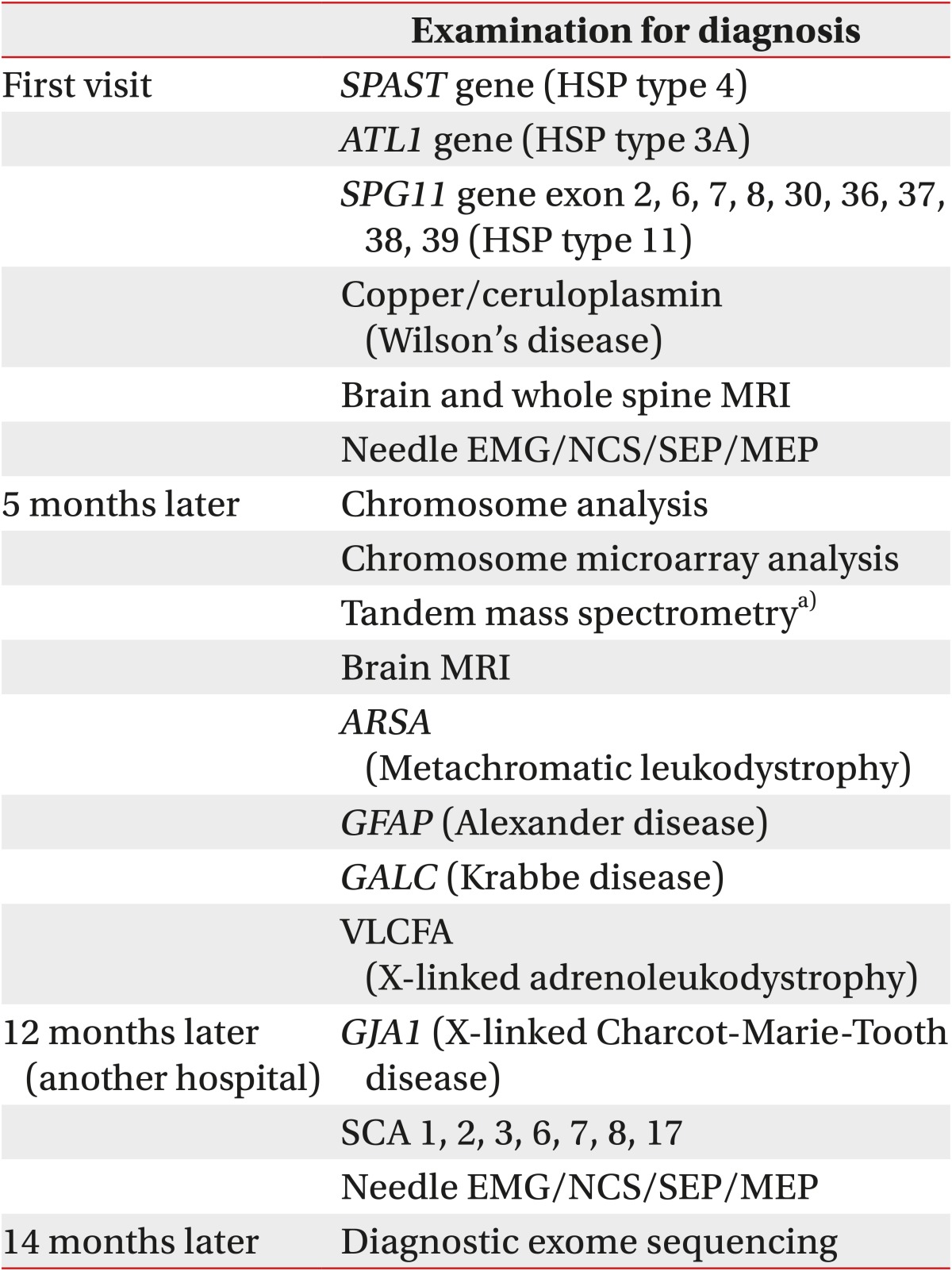
Table 2
Information on diagnosis of the patient's disease for the past 14 months before undergoing CES
CES, clinical exome sequencing; HSP, hereditary spastic paraplegia; MRI, magnetic resonance imaging; EMG, electromyography; NCS, nerve conduction study; SEP, somatosensory evoked potential; MEP, motor evoked potential; SCA, spastic cerebral ataxia; VLCFA, very long chain fatty acid.
a)Tandem mass spectrometry was performed for analysis of metabolic diseases, such as amino acid metabolic disorders, organic aciduria, and fatty acid metabolic disorders.
![]()
Go to :
 XML Download
XML Download