

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase (GNE) distal myopathy, which is also called Nonaka distal myopathy or distal myopathy with rimmed vacuoles or hereditary inclusion body myositis, is a late-adult onset autosomal recessive myopathy that is clinically characterized by progressive distal leg atrophy and weakness, especially anterior compartment of the lower limbs but sparing quadriceps group [1]. It is known that GNE myopathy patients become full-time wheelchair users within an average of 12 years after disease onset. The distal myopathy is caused by mutations in GNE gene (9p13.3), encoding a bi-functional protein with 2 enzymatic activities: Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase [2]. Molecular genetic analysis is becoming more and more important for the accurate diagnosis of GNE myopathy. More than 100 mutations in the GNE gene of different ethnic origin are reported to date [3].
We reported the genotype-phenotype correlation of certain compound heterozygous mutations i.e., the GNE gene, c.1714G>C (p.Val572Leu) and c.527A>T (p.Asp 176Val) for the first time in Korea.
CASE REPORT
Case description
A 48-year-old female visited Seoul National University Hospital because of left foot drop since 2 years prior. She was previously healthy without significant past medical history, except uterine myomectomy at the age of 42 years and breast benign tumorectomy in at the age of 45 years. The patient had no familial history of weakness or leg atrophy.
She had a mild gait disturbance at the age of 46 years and started to feel weakness on her left ankle dorsiflexor followed by right side weakness a year later. Serum creatine kinase was elevated at 404 IU/L and lactate dehydrogenase was within normal limits. A neurologic examination showed severe weakness of left ankle dorsiflexor (Medical Research Council [MRC] scale, grade 1) and long toe extensor (MRC scale, grade 2) with mild weakness of right ankle dorsiflexor (MRC scale, grade 3). Left achilles tendon reflex was reduced. However, neither muscle atrophy nor pseudohypertrophy was observed.
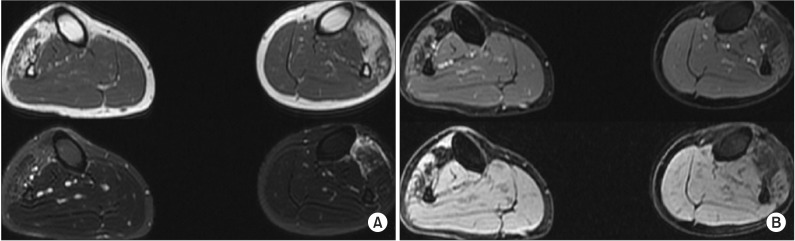
Electromyography (EMG) showed myopathic changes; short duration low amplitude polyphasic motor unit action potentials in early recruitment pattern that were found only in the muscles of the anterolateral compartment. Also, abnormal spontaneous activity was detected in the left tibialis anterior muscle. However, abnormal responses in the vastus medialis and gastrocnemius muscles were not observed. Nerve conduction studies revealed decreased amplitude of compound motor action potentials in the bilateral tibialis anterior muscles with no other abnormal finding. Magnetic Resonance Imaging (MRI) delineated diffuse fatty degeneration and atrophy of anterior and lateral compartment of the muscles of both lower legs (Fig. 1).
Muscle biopsy from left tibialis anterior muscle showed several rimmed vacuoles found in many degenerated and atrophic myofibers. There was marked size variation of myofibers that were mostly rounded in shape. Marked endomyseal fibrosis and fatty change were present, which were consistent with severe myopathic change. Also immunohistochemistry of desmin (1:200; Dako, Glostrup, Denmark) for myofibrillar myopathies, Titin (TTN, 1:200; Abnova, Taiwan) for Finnish (tibial) distal myopathy, and ZASP (1:200) for Markesbery distal myopathy showed findings within normal limits. Some degenerating myofibers were positive for desmin antibodies (Fig. 2).
Germ line mutation analysis: DNA extraction and sequencing
DNA was extracted from peripheral blood lymphocytes. First, the genomic sequence for ZASP exon 6 was conducted to rule out the initially suspected disease of Markesbery distal myopathy. However no mutation was detected. Next, all exons and splice regions of the GNE gene were sequenced and NG_008246.1 and NM_005476.5 were used as a reference sequence.
We identified a c.1714G>C/c.527A>T compound heterozygous mutation that were compound heterozygote of G-to-C transition at nucleotide position (c.1714G>C), which change an amino acid at codon 572 from valine to leucin (p. Val572Leu), and an A-to-T transition at nucleotide position (c.527A>T), which change an amino acid at codon 176 from aspartic acid to valine (p.Asp176Val) (Fig. 3).
DISCUSSION
c.1714G>C/c.527A>T compound heterozygous mutation is the second most frequent mutation of GNE myopathy in Japan. However, to the best of our knowledge, GNE myopathy due to c.1714G>C/c.527A>T compound heterozygous mutation has not been described in Korea. In this case, considering late symptom onset, predominantly anterior compartment involvement (asymmetric weakness of ankle dorsiflexor) and absence of family history (sporadic) highly suggested tibial muscular dystrophy (Udd myopathy), ZASPopathy (Markesbery-Griggs late onset distal myopathy), desminopathy and GNE myopathy (Nonaka myopathy, distal myopathy with rimmed vacuoles). Owing to its atypical and mild clinical feature, GNE myopathy with c.1714G>C/c.527A>T compound heterozygous mutation may produce confusion in clinical diagnosis. The combination of clinical features and ancillary studies such as electrodiagnostic studies, muscle biopsy and muscle imaging can narrow the range of causative genes for a certain distal myopathy and recent rapid advance in molecular genetic studies helps to confirm the diagnosis of distal myopathies [4].
As there are only a few single case reports or relatively small case series regarding GNE myopathy, it is difficult to determine genotype-phenotype correlation. However, recently Cho et al. [5] reported a single-ethnic cohort of GNE mutation composed of 212 patients with GNE myopathy originating from 201 Japanese families. The results from their study indicated that patients with homozygous c.1714G>C mutations had early symptom onset (23.9±7.1 years, p<0.01) and the majority of wheelchair-bound patients belonged to this group. On the other hand, patients with c.1714G>C/c.527A>T compound heterozygous mutation developed symptoms at a later age (37.6±12.6 years, p<0.01), and there were no wheelchair-bound patients at the time of genetic analysis in this group [56]. In view of the phenotype and the genotype correlation, the homozygous mutations in the kinase domain cause well-known typical clinical features of GNE myopathy, but compound heterozygous mutations of GNE showed varied clinical features and disease course. The phenotype of our case is consistent with the conclusion of Cho et al. [5].
Only a few cases of GNE myopathy in Korea are reported; and c.1714G>C/c.527A>T compound heterozygous mutation was first detected and fully described in our case. Although it is still unclear how GNE mutations cause various defects in protein metabolism, the ongoing identification of how each specific GNE mutation results in its clinical and pathologic feature will contribute to our understanding including the clinical diagnosis and treatment.


 XML Download
XML Download