

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ullrich congenital muscular dystrophy (UCMD) is characterized by congenital weakness, hypotonia, proximal joint contractures, and striking hyperlaxity of distal joints. In contrast to other types of congenital muscular dystrophy (CMD), patients with UCMD show normal intelligence and normal brain structure. UCMD is basically due to a defect in extra cellular matrix protein, collagen type VI, that is critical for skeletal muscle stability, regeneration, and muscle cell matrix adhesion. It is an autosomal recessive (or more rarely dominant) disorder caused by a mutation in one of the 3 collagen type VI genes (COL6A1, COL6A2, COL6A3) [1]. We report a case of UCMD diagnosed with genetic testing, who shows clinical features that suggest collagen type VI related muscle disorder.
CASE REPORT
A 37-year-old woman who cannot walk independently visited our outpatient clinic. She was born at term by normal vaginal delivery following an uncomplicated pregnancy with apparently normal fetal movements. In developmental history, her motor developmental milestones were delayed. She was able to sit at 12 months, crawled without support at 16 months, and walked without support at 24 months. A local hospital informed the possibility of poliomyelitis, when the patient showed a waddling gait after achieving her independent bipedal locomotion. At the age of eight, though the patient showed equinus foot deformity, she was able to run and walk 200 m independently to school. At the age of 13, the equinus worsened and the gait became slower. The patient ended up walking with clutches due to progressive weakness of her lower legs. In her early 20s, walking up the stairs was possible, but it became difficult by her late 20s. Until then, the ability to sit and to stand made her sedentary life possible. But from her early 30s, she was mostly bed-ridden and wheelchair-dependent due to worsened weakness. There is no family history of similar condition. She has six sisters and one brother with normal gait. Her intelligence was normal, so no history was found for difficulty in academic activities. At the age of 34, she was admitted to a general hospital for diagnostic work up and for the management of insidious onset of dyspnea (pCO2 110 mmHg on arterial blood gas analysis [ABGA]), edema of lower legs, and orthopnea. In electrophysiologic study, there were no definite abnormalities on sensory and motor nerve conduction study of upper and lower extremities. Needle electromyography on biceps brachii, extensor carpi radialis, rectus femoris, and tibialis anterior showed short duration, as well as short and long polyphasic motor unit action potential (MUAP) on volition with early and complete MUAP recruitment. A muscle biopsy was performed on right vastus lateralis. There was infiltration of adipose tissue in many atrophic myofibers with central nuclei and endomyseal fibrosis. Immunofluorescence and electron microscopy were not performed. She was managed under the impression on congenital myopathy. Nocturnal home mask ventilator care was applied due to orthopnea, after being discharged from the hospital. At the age of 35, the patient had right femur neck fracture due to a motor vehicle accident, and underwent an open reduction and multiple pinning fixations.
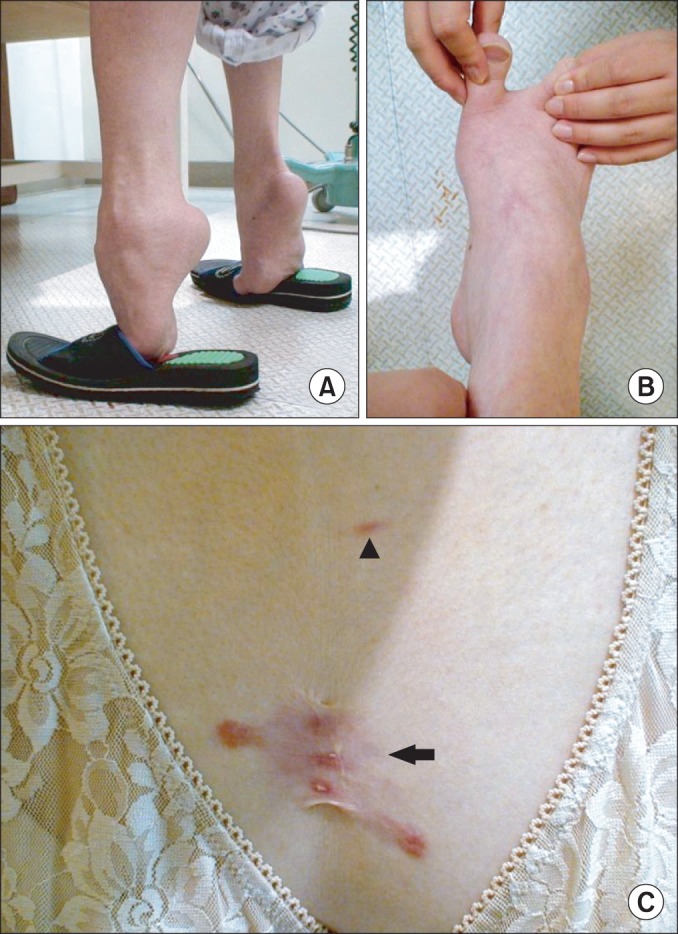
On physical examination, fixed hip flexion contracture (right/left, 20°/20°) and severe ankle plantar flexion (right/left, 50°/60°) contracture were noted. She walked with severe equinus with weight-bearing only at the metatarsal head area (Fig. 1A) and was able to walk independently with walker for a short distance (5 to 10 m). In addition, her toes were widely spread at standing. She was unable to run and had difficulty climbing stairs without assist. On neurologic examinations, functions of the sensory and cerebellar system were normal. The patient presented tightness in bilateral sternocleidomastoid muscles, long finger flexors, hamstrings, and Achilles tendon. She had right dorsolumbar scoliosis (Cobb's angle 38°), and range of motion of the spine was markedly limited in cervical and lumbosacral area. However, her distal foot joints showed excessive laxity and hypermobile toes (Fig. 1B). Motor system examination showed symmetrically atrophic upper and lower limbs without fasciculation. Proximal group of muscles had power of 4/5 and distal had 3/5, based on the Medical Research Council scale. Skin examination revealed soft velvety skin on the palms and the soles. Abnormal hypertrophic scarring with keloid formation was present on the incision site of the right thigh, and other keloids were formed on her chest, left shoulder, and intragluteal fold (Fig. 1C).
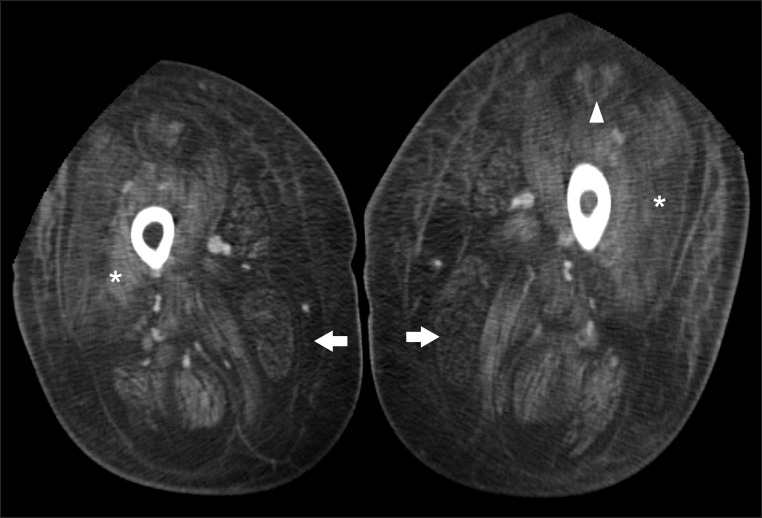
On laboratory examination, the serum creatine kinase level (33 IU/L) was in the normal range. The ABGA and the capnography represented chronic respiratory acidosis with hypoventilation (pH 7.3; pCO2 61.3 mmHg; pO2 59.9 mmHg; HCO3 33.6 mmol/L). The pulmonary function test showed severely restrictive respiratory pattern, in which the functional vital capacity is 0.39 L or 12% of the predicted value at supine position, and 0.84 L or 25% of the predicted value at sitting position. Results of cardiac evaluation including echocardiography were normal. Her hip computed tomography (CT) showed diffuse fatty infiltration and atrophy of thigh muscles in both sides, and 'central shadow' sign at both rectus femoris. This is the characteristic of muscle involvement pattern in UCMD, where the peripheral regions of the muscles are involved, while relatively sparing the central parts (Fig. 2).
According to the medical history, clinical manifestations include skin and musculoskeletal abnormalities that suggest collagen type VI related muscle disorders, in which, UCMD was highly suggested. In order to confirm the diagnosis, COL6A1 gene sequencing was performed in the next step, because the patient refused repeat of the muscle biopsy.
After obtaining a written informed consent from the patient and the family members of the patient, peripheral blood samples were obtained. Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. All 35 coding exons and their flanking intronic regions of COL6A1 were amplified by polymerase chain reaction using gene-specific primers on a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA). Sequencing was performed using the same primers on the ABI Prism 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA) using the BigDye Terminator Cycle Sequencing Reaction Kit (Applied Biosystems). The sequences obtained were compared with the reference sequence of COL6A1 gene (NM_001848.2) with Sequencer ver. 4.10.1 software (Gene Codes Corporation, Ann Arbor, MI, USA).
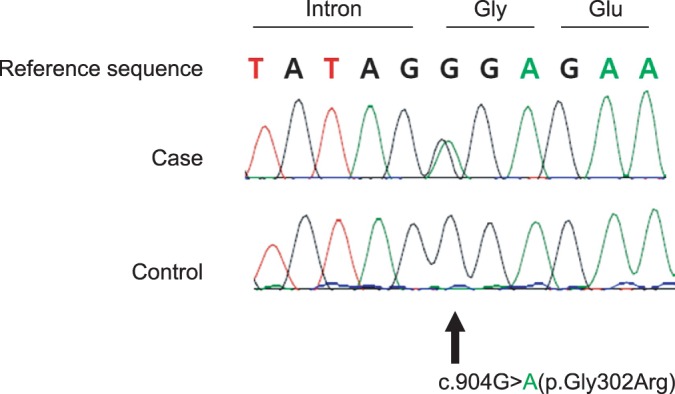
As a result, we found three variants of unknown significance; one homozygous intronic variant (c.588+19dupC), one heterozygous missense variant in the start of exon 11 (c.904G>A; p.Gly302Arg), and one homozygous silent variant (c.1095T>C; p.Gly365=). Of those variants, c.904G>A (p.Gly302Arg) was considered as a potential candidate for disease-causing mutation (Fig. 3). This is a novel mutation that has never been reported before. Therefore, additional sequencing analysis for the c.904G>A variant was done, and it revealed that her mother and her sister, who had no symptoms, did not have the variant. Based on these results, we diagnosed the patient as UCMD, with novel c.904G>A (p.Gly302Arg) variant.
The patient was placed on the volume control ventilator, because the results of ABGA still showed chronic respiratory hypercapnic acidosis with metabolic compensation (pCO2 61.3 mmHg), even after using nocturnal bilevel positive airway pressure (BiPAP). Symptom relief was observed (pCO2 36.3 mmHg) in the serial ABGA, and she was discharged from the hospital after setting the volume control mode (tidal volume 750 mL; respiratory rate 12/min).
DISCUSSION
Mutations in the genes encoding collagen type VI (COL6A1, COL6A2, and COL6A3) cause Bethlem myopathy (BM) and UCMD. These two conditions show variable combinations of muscle wasting and weakness, joint contractures, distal laxity, and respiratory compromise. BM is a relatively mild dominantly inherited disorder characterized by proximal weakness and distal joint contractures. UCMD was originally described as an autosomal recessive condition, causing severe muscle weakness with proximal joint contractures and distal hyperlaxity [2]. In collagen type VI related muscle disorders, BM and UCMD can be differentiated if immunohistochemical staining is done to collagen type VI, because BM mostly seems normal and UCMD is not stained or decreased [3]. However, recently many cases have been reported that make this differentiation ambiguous, and suggestions are made that the two diseases should be interpreted in one spectrum [4]. Orthopedic deformities (joint contractures, scoliosis) and respiratory impairment with diaphragmatic failure of UCMD generally develop within the first decade of life, and may be life-threatening. Arrest of motor milestones with no acquisition of walking ability is seen in a subset of patients. Most other children are able to walk but they will show progression of muscle weakness with loss of ambulation at about 10 years of age. In their late childhood or young adulthood, they will require mechanical ventilation. Dermatologic manifestations, such as follicular hyperkeratosis and hypertrophic scars or keloid formation, are common. Other common findings include normal cognitive abilities, normal or only slightly raised serum creatine kinase levels, and absence of cardiac phenotype [5].
The patient of our case had scoliosis, joint contracture, distal joint hyperlaxity, difficulty of respiration, and many skin keloids. Her hip CT showed diffuse fatty infiltration of thigh muscles with relative sparing of the central part of the muscles in the anteromedial thigh. The pattern of muscle involvement shows a similar appearance with UCMD patients' recent magnetic resonance imaging study of the lower extremities [6]. The muscle imaging study is not performed on a regular basis in the diagnostic process of neuromuscular disorders, because the interpretation of the scans is perceived as requiring a high level of expertise. However, it could be used in the selected patients, as an additional tool in the differential diagnosis of muscle disorders. The clinical and radiologic features of the patient including abnormalities in muscle, bone, joint, and skin made us suspect collagen type VI related disorders. In the diagnosis of UCMD, it is important to consider not only the clinical features of myopathy, such as gait disturbance and muscle weakness, but also need to consider the orthopedic deformity, dermatologic manifestation, and radiologic findings.
Traditionally, the diagnosis of CMD had been relied on clinical findings, brain and muscle imaging, muscle biopsy histology, and muscle and/or skin immunohistochemical staining [7]. Immunohistochemical study in muscle biopsy is a very expensive laboratory test, and it may be difficult to perform this routinely in hospitals that lack the diverse antibodies for cellular material. With the expanding role of molecular genetic testing in confirming the diagnosis of a CMD subtype, this case showed an example of applying molecular genetic tests for a patient with clinically suspicious UCMD, without immunohistochemical study of muscle biopsy.
In this case, sequencing of COL6A1 gene revealed a missense variant (c.904G>A; p.Gly302Arg) which was considered as disease-causing mutation. G to A substitution in the locus 904 reads to the substitution of 302nd glycine to arginine. Because glycine sequence have important role in structure and function of collagen protein, substitution of glycine with another amino acids could give impact on collagen. Therefore this missense variant (c.904G>A; p.Gly302Arg) is considered as a candidate for disease-causing mutation [8]. Further investigation is required to confirm whether this novel variant is responsible for such phenotype. In the previous Korean case of UCMD, a known mutation (c.868G>A; p.Gly290Arg) was reported in triple helix domain [9]. The mutation found in our case is novel, which is mutated in a different site compared to the previous reports.
If musculoskeletal and dermatologic manifestations with the findings of radiologic study imply abnormalities in collagen type VI network, COL6A related CMD should be suspected. With the expanding role of molecular genetic testing to confirm the diagnosis of a CMD subtype, molecular genetic tests should be considered for a patient with clinically suspicious UCMD when proper immunohistochemical studies in muscle biopsy are not available.
 XML Download
XML Download