

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The corpus callosum is the largest interhemispheric connective structure in the brain [1]. Anomalies of the corpus callosum are observed in a variety of conditions that disrupt early cerebral development, including chromosomal and metabolic disorders, as well as intrauterine exposure to teratogens and infection [2]. Recently, a gene defect, haploinsufficiency of AKT3, was also reported, which can be a cause of microcephaly and agenesis of the corpus callosum [3]. Callosal anomalies are often accompanied by other central nervous system (CNS) and/or somatic anomalies [4]. Szabo et al. [1] studied the birth prevalence and clinical spectrum of corpus callosal anomaly patients in Hungary. Glass et al. [2] researched the prevalence and demographic risk factors of callosal anomaly in patients over 20 years in California. They reported that patients with corpus callosal anomalies had other associated CNS and/or somatic anomalies, and more severe callosal anomaly was associated with more functional impairment.
Despite the strong possibility of differences due to ethnicity, in comparison with prior reports, studies of corpus callosal anomaly in Korea have focused on prenatal diagnosis [5], and the clinical outcomes of patients with callosal anomaly have not been addressed. Therefore, the purpose of this study was to describe the clinical features of corpus callosal anomalies in children and adolescents, and to evaluate the developmental prognosis of those patients in Korea.
Go to : 
CASE REPORT
This study was a retrospective review of medical records of children and adolescents with corpus callosal anomalies in our hospital. The subjects were patients who visited our hospital and underwent brain magnetic resonance imaging (MRI), from November 2005 to January 2012. The diagnosis of corpus callosal anomaly was established by MRI, and expert radiologists reviewed the images. We excluded patients with extensive CNS anomalies (e.g., lissencephaly, holoprosencephaly, or bilateral schizencephaly), because the corpus callosal anomaly can be secondary to these CNS anomalies [1]. For the same reason, preterm infants with a thin corpus callosum accompanied by periventricular leukomalacia were also excluded.
Forty-five patients (30 males and 15 females; age ranging from 1 to 17 years) were reported to have agenesis/hypoplasia of the corpus callosum. On the basis of accompanying CNS or somatic anomalies, patients were classified into four groups, as follows (Fig. 1): group 1, isolated agenesis (complete or partial) or hypoplasia of the corpus callosum; group 2, agenesis (complete or partial) or hypoplasia of the corpus callosum, associated with other CNS abnormalities; group 3, agenesis (complete or partial) or hypoplasia of the corpus callosum, associated with both CNS and somatic abnormalities; and group 4, agenesis (complete or partial) or hypoplasia of the corpus callosum, associated only with somatic abnormalities.
 | Fig. 1Classifications of callosal anomalies. Group 1, isolated agenesis/hypoplasia of the corpus callosum; group 2, agenesis/hypoplasia of the corpus callosum in association with other CNS abnormalities; group 3, agenesis/hypoplasia of the corpus callosum in association with both CNS and non-CNS abnormalities; and group 4, agenesis/hypoplasia of the corpus callosum in association with non-CNS abnormalities. CNS: central nervous system.
|
In these classifications, complete agenesis means total absence of the corpus callosum, and partial agenesis means partial absence of the corpus callosum. Hypoplasia was defined as when the corpus callosum is fully formed, but is thinner than expected for the age of the individual, according to the definition of previous reports [1,6], which infers the presence of fewer axons.
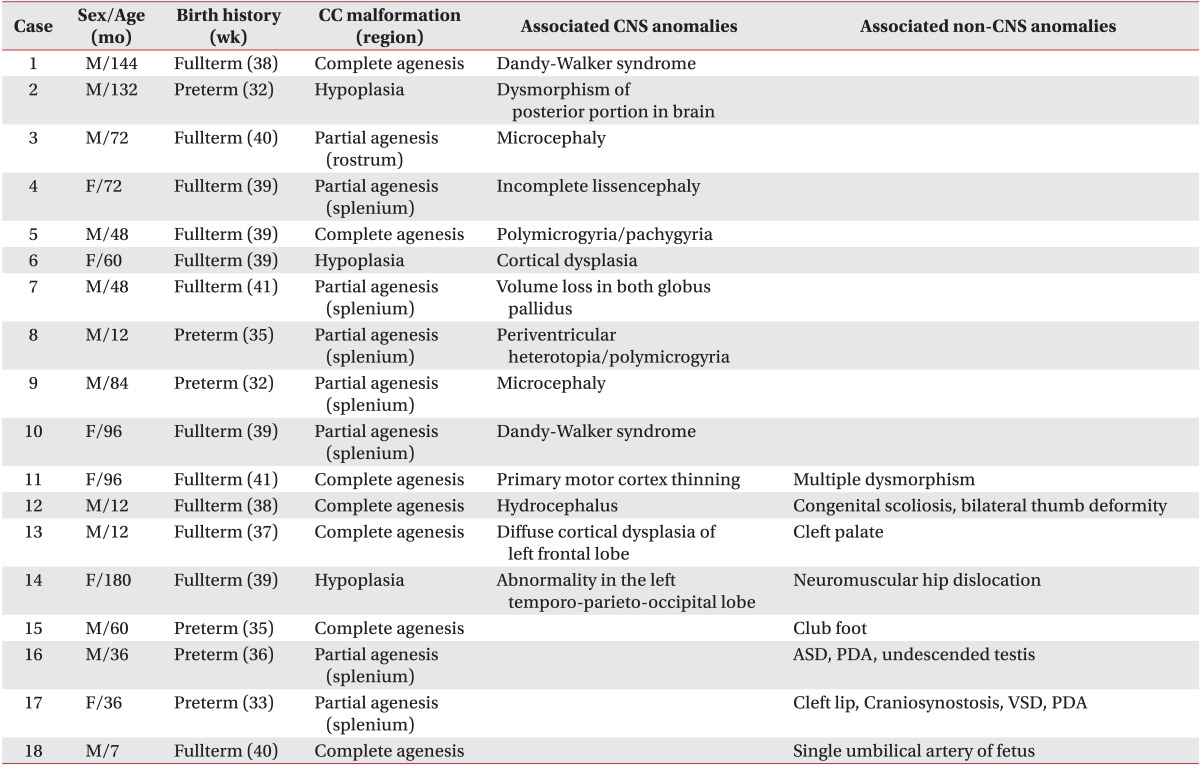
Among these 45 patients, 31 patients visited our rehabilitation department, and the distribution of patients into the four groups was as follows: 13 patients in group 1; 10 patients in group 2; four patients in group 3; and four patients in group 4. We reviewed patient medical records, and the clinical features were classified into cerebral palsy (CP), motor delay, intellectual disability, and normal development, in each group of anomaly (Fig. 2). The diagnosis of CP was made by experienced pediatric physiatrists, based on the brain MRI, and clinical features, such as paralysis, spasticity, etc. The children with motor delay meant the people who were not diagnosed with intellectual disability, and did not show any neurologic symptoms and signs suggesting CP, but who did show delayed motor milestones. The intellectual disability was defined as below 70 in IQ test that was executed before 18 years old, and impairment in cognitive function and social adjustment. With the exception of 13 patients with an isolated corpus callosal anomaly, 18 patients with CNS or non-CNS abnormalities had various concomitant anomalies or syndromes (Table 1).
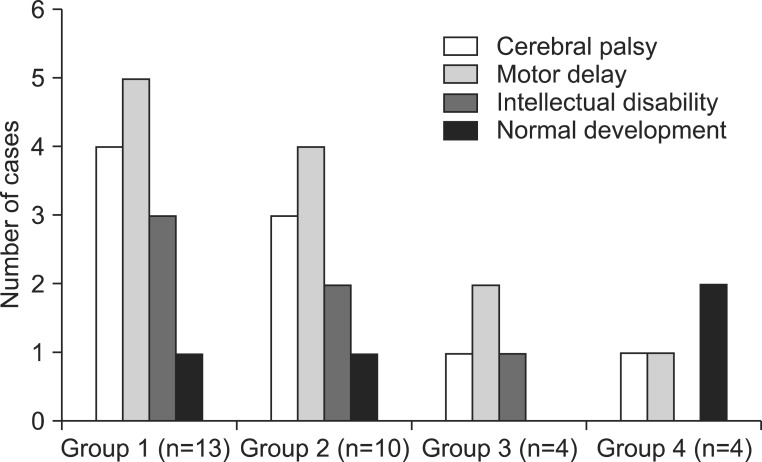
Among the 31 patients who visited the rehabilitation clinic, nine were diagnosed with CP, and four developed normally, without impairments in motor or cognitive functions. In the four patients with normal development, the first case visited the department of rehabilitation due to both equinus and isolated corpus callosal hypoplasia detected in a brain MRI at 3 years of age, and this case was classified as group 1. However, the equinus spontaneously disappeared during follow-up. The second case was born with neonatal hyperbilirubinemia at 38 weeks gestation. After being in the intensive care unit for 2 weeks, he was brought to the department of rehabilitation for a routine check-up, and was found to have corpus callosal agenesis, polymicrogyria, and pachygyria on brain MRI at 4 years old, and was classified as group 2. The other two cases were classified as group 4. One case was born prematurely at 35 weeks, and was brought to the rehabilitation clinic due to club foot deformity. In this case, the callosal anomaly was observed on brain MRI at 5 years old. The other case had a single umbilical artery at birth, and was brought to the rehabilitation clinic for a routine check-up at 7 months old, at which time callosal agenesis was observed on brain MRI.
Five of nine patients with CP (three in group 2, one in group 3, and one in group 4), accompanied by other CNS and/or somatic anomalies, showed worse scores on the Gross Motor Function Classification System (GMFCS; all patients were GMFCS level V), compared with the four patients with isolated callosal anomaly (two patients with GMFCS level II, and two patients with GMFCS IV).
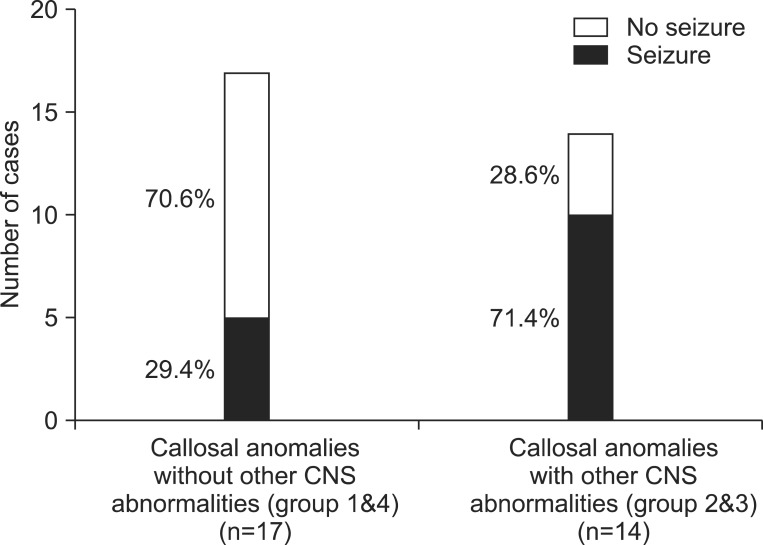
Patients with anomaly of the corpus callosum have a high seizure risk in general, and patients with other CNS anomalies also have a higher seizure risk. Among the 17 patients in groups 1 and 4 who had no other CNS anomaly, five patients (four patients in group 1 and one patient in group 4) had a clinical history of seizures, which is a rate of about 29.4%. Among the 14 patients in groups 2 and 3 with accompanying CNS anomalies, 10 patients (eight patients in group 2 and two patients in group 4) presented with a history of seizures, which is a rate of about 71.4% (Fig. 3).
Go to :
DISCUSSION
The purpose of this study was to describe the clinical features of children and adolescents with corpus callosal anomalies, from the aspect of combined clinical characteristics, and to evaluate the developmental characteristics of those who visited our hospital.
The proportion of patients with isolated agenesis of the corpus callosum that showed normal development was 19 out of 26, in a previous study by Mangione et al. [7]. However, in the present study, only one out of 13 cases of isolated agenesis or hypoplasia of the corpus callosum developed normally. This difference may be caused by the use of different definitions of normal development. Mangione et al. [7] defined delayed development as 79 or less on the Development Quotient calculated from Child Developmental Inventory (DQ-CDI), and some patients with convulsion, hypotonia, and strabismus were included in the normal developmental group. However, the definition of normal development in this study was stricter, in that the absence of developmental delay, intellectual disability, abnormal neurologic findings, and epilepsy was required. A previous study by Szabo et al. [1] that applied criteria similar to that of this study reported that 13 out of 18 patients with isolated agenesis or hypoplasia showed developmental delay, intellectual disability, or epilepsy. Another possibility explanation for the low rate of normal development in this study is that we only recruited subjects who visited the rehabilitation clinic in our hospital; not patients with incidental corpus callosal anomaly. Therefore, the results of this study should not be considered to be representative of the prevalence in the general population of Korea.
In patients with CP in this study, other accompanying CNS and/or somatic anomalies showed worse effects on gross motor function, as measured by GMFCS, compared with isolated callosal anomaly. A previous study by Tang et al. [8] also reported that patients with accompanying abnormal sulcal morphology, cerebellar abnormalities, vermian abnormalities, and brain stem abnormalities showed poor neurodevelopmental outcomes. They analyzed the causal relationship of structural anomaly in fetal MR images with functional impairments, and reported that patients with abnormal sulcal morphology and infratentorial abnormalities had poor outcomes. Our study did not try to elucidate the relationship between specific brain structure, and the function of patients with CP. However, patients with other associated CNS abnormalities had worse levels of gross motor function.
Considering the seizure risk, patients with other accompanying CNS lesions (groups 1 and 4) had a higher seizure risk, compared with those without CNS lesions (groups 2 and 3), in this study.One previous study reported that none of six children with isolated agenesis of the corpus callosum had seizures, while all three children with other CNS lesions demonstrated early seizures [9]. In contrast, Byrd et al. [10] reported that 8 out of 26 cases with isolated corpus callosal agenesis presented with seizures. This seizure risk is higher than that in the general population, which is commonly reported as 4 to 8 per 1,000 people. Therefore, the higher seizure risk in cases of corpus callosal anomaly should be taken into account; and it should be noted, in particular, that this risk is much higher in cases with other associated CNS anomalies, compared with patients with isolated callosal anomaly. Additionally, seizure control is difficult in patients with CNS lesions, and severe developmental delay is often associated with these conditions.
In conclusion, we demonstrated the clinical characteristics of corpus callosal anomalies in Korea. Patients with CP accompanied by other CNS and/or somatic anomalies showed worse levels of GMFCS, compared with those with isolated callosal anomaly. In addition, patients with other CNS anomalies also had a higher seizure risk.
This study has limitations of small sample size, and selection bias. The results of this study should not be generalized to the Korean population, because we analyzed only subjects who visited our rehabilitation clinic. Further studies are required, to prospectively analyze clinical findings in the general population. In addition, we could not focus on the clinical features of corpus callosal anomaly itself, but dealt with the various accompanied anomalies, and their clinical characteristics. The clinical outcomes of isolated corpus callosal anomaly have been reported variously, and the reason for the variety has not yet been elucidated. Therefore, the mechanism of how this anomaly affects the neurologic functions in the human brain should be studied in further researches.
Go to :
 XML Download
XML Download