

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemifacial spasm (HFS) is characterized by spontaneous, paroxysms of rapid and irregular clonic twitching of facial muscles that occur on one side of the face. This abnormal activity usually originates in the muscles around the eyes and eventually spreads throughout the hemifacial muscles. HFS is a consequence of chronic subclinical facial nerve damage. Many researchers have demonstrated the origin of such facial spasms as a vascular compression of the nerve at its root exit zone, although the compressing vessel is not always found [1-3]. Neuroimaging studies, which frequently reveal contacts between the nerve and the offending vessel, and histological evidence of demyelination or nerve degeneration at the root exit zone also support the theory that HFS is caused by compression of the facial nerve. Furthermore, clinical improvement after surgical intervention at the posterior fossa has been reported [4-6]. However, there is also evidence that facial motor neurons are hyperexcitable in patients with HFS [7-10]. Therefore, extrinsic irritation of the facial nerve at the posterior fossa likely generates antidromic inputs to facial motor neurons, thereby causing changes in excitability and spontaneous or reflex firing of motor neurons [5,11,12].
The diagnosis of idiopathic HFS is usually clinical; however, some patients do not exhibit hemifacial twitching on examination. In this situation, electrophysiological testing may help distinguish HFS from abnormal facial movements [13-15]. The characteristic electrophysiological abnormalities observed in HFS is considered an additional response of the orbicularis oris muscle to application of electrical stimuli to the supraorbital nerve. Normally, the trigeminofacial reflex should be limited to the orbicularis oculi muscle [6]. Two possible pathophysiological mechanisms may underlie the abnormal orbicularis oris response. In the peripheral theory, the electrical stimulus activates the supraorbital nerve afferents that reach the facial motor neurons of the orbicularis oculi muscles, but the facial nerve axons to the orbicularis oculi cause a lateral spread of excitation to the axons innervating the orbicularis oris at the site of presumed nerve demyelination [1,6,16,17]. In the alternative central theory, the supraorbital stimulus activates not only the facial motor neurons that innervate the orbicularis oculi muscle, but also those that innervate the orbicularis oris muscle because of increased facial motor neuronal excitability, facial nucleus reorganization, or even interneuronal hyperexcitability [5,11,12].
In patients with HFS, the lateral spread response can be elicited by electrical stimulation of one branch of the facial nerve and can be recorded electromyographically from muscles that are innervated by other branches of the nerve [1]. These responses also occur because of axon-axon ephaptic transmission or facial motor neuronal hyperexcitability [18].
Whether the lateral spread response originates from a central or peripheral site is debated. We evaluates the pathophysiological mechanism of HFS based on electrophysiological examinations, such as supraorbital nerve stimulation and lateral spread tests, by suppressing the patient's central nervous system (CNS) via intravenous administration of diazepam.
We hypothesized that the orbicularis oris muscle response in supraorbital nerve stimulation or lateral spread tests would be altered by diazepam. We performed these two tests in a small cohort of patients with HFS during CNS suppression using intravenous administration of diazepam. We speculated that the orbicularis oris muscle response elicited by supraorbital nerve stimulation would be delayed because it would pass through the facial motor neuron. However, in the lateral spread test, we reasoned that if orbicularis oris muscle responses were caused by ephaptic transmission between axons there should be a constant latency of these responses as they are elicited by the facial motor neuron. Conversely, if these responses were caused by facial motor neuronal hyperexcitability, the latency of the orbicularis oris muscle response in the lateral spread test would be delayed because responses were elicited from the facial motor neuron.
MATERIALS AND METHODS
Subjects
Six patients with HFS who met the inclusion criteria detailed below were prospectively enrolled in this study after providing informed consent. Ethics approval for our analyses was obtained from the Institutional Review Board of our institution. The patient's inclusion criteria were as follows: 1) age >18 years; 2) a clinical diagnosis of HFS, such as paroxysmal and repetitive involuntary hemifacial contractions of the muscles that control facial expression, which were confirmed by a neurologist, neurosurgeon, or physiatrist; and 3) involuntary spasms extending to the orbicularis oris muscle.
Patients were excluded based of the following criteria: 1) presence of a movement disorder, such as tics, dystonia, hemimasticatory spasm, focal seizure, or blepharospasm; 2) presence of a degenerative brain lesion; 3) history of facial neuropathy; 4) botulinum toxin treatment within the previous year; 5) treatment with CNS suppressants, such as benzodiazepine within the previous 6 months; 6) prior brain surgery; or 7) problems with comprehension that might affect the study findings.
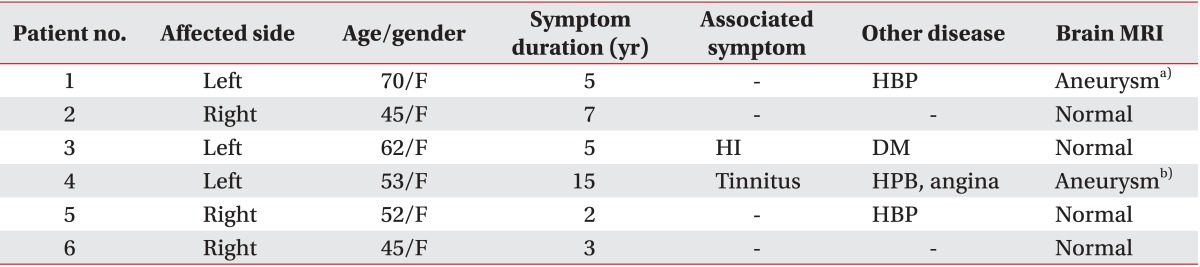
The duration of symptoms in our patients was 2-15 years. Spasms affected both the right and left side of the face in three patients. The clinical intensity of the spasms varied, ranging from an occasional abnormal twitching to full-blown spasms lasting for several seconds. None of our patients had a personal or family history of any other movement disorder or previous facial palsy. One patient was treated with botulinum toxin three times; however, our electrophysiological tests were performed 1 year after her last treatment. Magnetic resonance imaging findings of the posterior fossa were normal in four patients. One patient had a large aneurysm of the cavernous and ophthalmic segment of the left internal carotid artery, and another had a small medial directed aneurysm at the supraclinoid segment of the left internal carotid artery. Patient characteristics are presented in Table 1.
Methods
Electrophysiological analyses
Facial nerve conduction and blink reflex studies were performed using an electromyography instrument Medelec Synergy (VIASYS Healthcare, Surrey, UK). Disposable 4-disc electrodes with leads (019-400400 Nicolet, VIASYS Healthcare) were used as the active, reference, and ground electrodes. We maintained skin temperature >32℃ and used disposable alcohol cotton swabs for skin preparation. All studies were performed while the subject was lying quietly on a couch in a supine position. The ground electrode was placed on the chin, and electrical stimuli were delivered using a surface stimulator (Deluxe Bipolar Stimulator 031K074, VIASYS Healthcare). All stimuli were delivered when the recording muscles were at rest. Further testing was not performed until complete relaxation when spasms appeared after electrical stimulation.
Facial nerve conduction study
Active surface disc electrodes were placed on the bilateral frontalis, orbicularis oculi, nasalis, and orbicularis oris muscles, and a reference electrode was applied on the contralateral side of the same muscle [19]. Constant current stimuli for 0.1 ms were applied at a frequency of 1 Hz (or slower) to achieve supramaximal activation of the facial nerve at the tragus. We measured onset latency and peak-to-peak amplitude of the compound muscle action potential, which was recorded from the facial muscles after stimulation of the facial nerve on both sides.
Blink reflex
Active surface disc electrodes were placed at the midpoint of both inferior orbicularis oculi muscles, and the references were placed laterally to the orbit using dual channel recording. The cathode of the stimulator was applied to the supraorbital notch, whereas the anode was placed in the superolateral position. Constant current paired stimuli of 0.1 ms in duration at an intensity of 20-30 mA, were delivered at a frequency of 1 Hz (or slower). Ten stimuli were applied to each side, and the corresponding traces, which were recorded at a sweep speed of 100 ms and sensitivity of 500 µV, were stored for offline analyses. The latencies of the R1, R2, and contralateral R2 were read, and the shortest values of the blink reflex latencies were obtained.
Orbicularis oris muscle response to supraorbital nerve stimulation
Active surface disc electrodes were placed on the midpoint of the inferior orbicularis oculi muscle for supraorbital nerve stimulation, and reference electrodes were placed laterally to the orbit on the symptomatic side. Another active surface disc electrode was placed on the inferior orbicularis oris muscle with its reference surface disc electrode placed laterally to the lips on the symptomatic side using dual channel recording. Electrical stimulation was applied in the same manner as that used for the blink reflex study described above. Ten stimuli were applied to the symptomatic side. Orbicularis oculi muscle responses were obtained in the same manner as that used in the blink reflex study. The responses were called R1 and R2 responses. Orbicularis oris muscle responses elicited by supraorbital nerve stimulation were obtained from orbicularis oris muscles. Latencies in the R1, R2, and orbicularis oris muscle responses were read, and the shortest values for these latencies were obtained.
Lateral spread test
Active surface disc electrodes were placed on the orbicularis oculi muscle with their reference surface disc electrodes placed on the contralateral orbicularis oculi muscle. An additional active surface disc electrode was placed on the orbicularis oris muscle with its reference surface disc electrode placed on the contralateral orbicularis oris muscle on the symptomatic side of the face by using a dual channel recording. Electrical stimulation was delivered to zygomatic branches of the facial nerve on the symptomatic side of the face. Constant current stimuli were applied for 0.1 ms at a frequency of 1 Hz (or slower) to achieve supramaximal activation of the nerve. Direct responses from the orbicularis oculi muscle and indirect responses from the orbicularis oris muscles were recorded simultaneously. The orbicularis oris muscle response to stimulation of the zygomatic branch of the facial nerve in HFS occurs with a latency of 8-10 ms [1,20], whereas the orbicularis oris muscle response to volume conduction of the facial nerve occurs with similar latency of the normal orbicularis oris muscle response to mandibular branch of facial nerve stimulation. Hence, to rule out volume-conducted orbicularis oris muscle response, we recorded the response when the latency of the orbicularis oris muscle response was >7 ms. In addition, to avoid volume conduction of the orbicularis oris muscle response, we carefully monitored the contraction of other facial muscles, such as the nasalis muscle, and we started the exam with lower stimulus intensity, then we gradually increased intensity of stimuli to achieve supramaximal activation. At least 10 responses of the same form and latency were recorded from each patient. Latencies in the initial deflections of the responses were measured. Sweep speed was 2 ms/division, and other recording parameters were the same as described above.
Experimental procedures
Electrophysiological studies were performed at 10 and 20 minutes after the subjects had received 10 mg of diazepam intravenously. The lateral spread test and orbicularis oris muscle response to supraorbital nerve stimulation were performed using the recording parameters described above. We monitored patients' blood pressure, heart rate, and oxygen saturation levels during the analysis to detect any possible risk of respiratory depression.
RESULTS
Facial nerve conduction and the blink reflex study
No difference in the compound motor action potential amplitude was observed between the asymptomatic and symptomatic sides of the face in any patient. We observed normal latencies for the R1, R2, and contralateral R2 reflexes in the blink reflex study.
Results obtained prior to diazepam injection
A direct response was obtained in the orbicularis oculi muscle upon stimulation of the zygomatic branch of the facial nerve. An indirect response was obtained in the orbicularis oris muscle upon stimulation of the zygomatic branch of the facial nerve. R1 and R2 responses were obtained in the orbicularis oculi muscles upon stimulation of the supraorbital nerve. Early and late responses were obtained in the orbicularis oris muscles upon stimulation of the supraorbital nerve.
We observed a response from the orbicularis oris muscle in all patients after electrical stimulation over the supraorbital nerve (Fig. 1B). We identified two different components of this response: an early short-duration stable component (early response) and a longer latency rather variable component (late response). Indirect responses by the orbicularis oris muscle of all patients were also apparent after stimulation of the zygomatic branch of the facial nerve (Fig. 1A).
Results obtained after diazepam injection
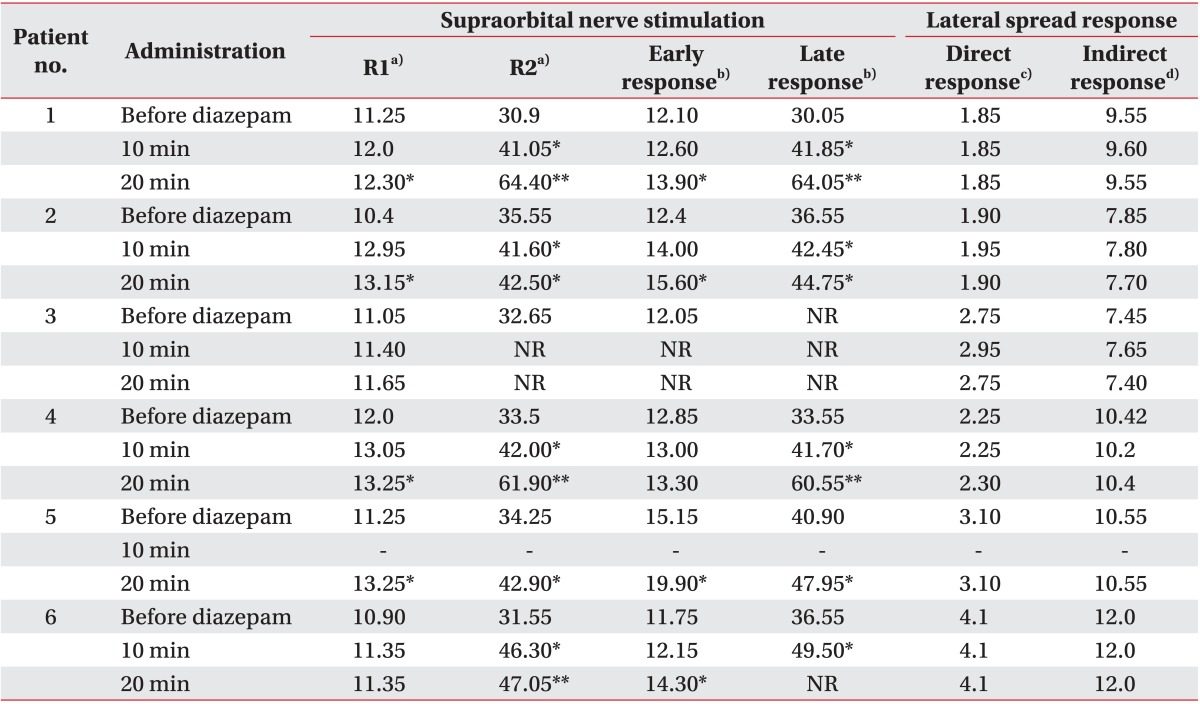
Table 2 lists the measured latencies of the direct and late responses elicited by stimulating the zygomatic branch of the facial nerve, and the latencies of the orbicularis oculi responses (R1 and R2 responses) and orbicularis oris muscle responses (early and late responses) after stimulation of the supraorbital nerve.
Orbicularis oris muscle responses to supraorbital nerve stimulation
Latencies of the ipsilateral R1, R2, early and late responses tended to increase over the course of the study period. This was particularly true for the R2 and late responses, which were significantly delayed (Fig. 2C, D). Four of six patients exhibited a 10% delay in the latencies of the R1 response and early response, 20 minutes after diazepam administration, as compared to their responses before diazepam administration. In the remaining two patients, although the latency delay was <10%, their latencies still tended to increase over time. Three of six patients had a >30% delay in the latencies of R2 responses 20 minutes after diazepam administration, and two patients had a >30% delay in the latencies of late responses after diazepam administration. One patient did not have any early or late responses after diazepam injection. This was possibly because of excess facial motor neuronal suppression occurred after intravenous administration of diazepam.
Lateral spread test
The latencies of the direct and indirect responses were consistent over time in all patients. (Table 2; Fig. 2A, B).
No major complications, such as respiratory depression, were observed in any of the subjects at anytime during the study. Patients reported drowsiness as their main complaint after diazepam administration.
DISCUSSION
Orbicularis oris muscle responses to supraorbital nerve stimulation or lateral spread responses in patients with HFS can be caused by two pathophysiological mechanisms. In the first mechanism, a lateral axon-axon spread of excitation in the facial nerve fibers occurs after facial motor neuron activation [6,18,21]. In the second mechanism, enhanced excitability of the facial motor neurons that innervate the orbicularis oris muscle is enhanced, which are abnormally activated by inputs directed selectively to other facial motor neurons [12,22]. Several reports support the central theory of the origin of the lateral spread response in the "hyperexcitability of the facial nucleus" [7,8,20,21,23,24]. Some investigators have proposed that orbicularis oris muscle responses are facial F-responses and are enhanced in patients with HFS [21,23]. Ishikawa et al. [7] examined F-waves both pre- and post-operatively and during surgery and found evidence that supports this central theory. Their results, which demonstrated that F/M-wave amplitude ratios are correlated with the F/M-wave amplitude ratios of the lateral spread response, led them to conclude that the origin of enhanced F-waves is the same as that of the lateral spread response. The F-wave exhibits variable latency and amplitude because of the hyperexcitability of the central nucleus [23,24]. The same concept can be derived from the work of Moller [20], who determined the response latencies intraoperatively and proposed a nuclear origin for the activity.
Therefore, we inhibited the facial motor nucleus using diazepam to test the central origin hypothesis in the present study. Diazepam is a CNS depressant that operates through gamma aminobutyric acid (GABA) receptor inhibition. Cruccu et al. [25] studied blink reflexes in six healthy volunteers after intravenous administration of 10 mg diazepam. In that study, R1 was slightly suppressed, whereas the R2 reflex was deeply suppressed [25]. The action of diazepam as a CNS depressant at the brainstem may explain this observation [26]. Diazepam is effective for treating several forms of muscle hyperactivity, including spasticity, and acts as a suppressor of tendon and H-reflexes [27]. This muscle-relaxant action is considered secondary to the inhibition of both motor neurons and interneurons via the same receptors as those used by GABA [26]. Suppression of the R2 reflex is possibly associated with its polysynaptic circuit via the reticular formation and to the descending control of the circuit via corticoreticular projections. The reticular formation, where GABA and benzodiazepine receptors are coupled, is rich in GABA [28].
We measured the latencies of the R1 and R2 responses and of the early and late orbicularis oris muscle responses after supraorbital nerve stimulation. We observed a gradual response delay over time after the diazepam injection. In particular, the R2 response and late orbicularis oris muscle responses were delayed more markedly than was the R1 response or the early orbicularis oris muscle responses. The R1 and R2 responses and the early and late orbicularis oris muscle responses are signals elicited through the facial motor neuron in a supraorbital stimulation setting. Hence, the responses were delayed by diazepam-mediated suppression of facial motor neurons. Moreover, because of a polysynaptic circuit, the latency of the R2 response and late orbicularis oris muscle response after supraorbital nerve stimulation was delayed markedly. These findings suggest that diazepam suppresses facial motor neurons effectively.
If the facial nucleus is the site of abnormal cross transmission, then the orbicularis oris muscle response in a lateral spread test should be a waveform with delayed latency instead of a sustained response, as observed in supraorbital nerve stimulation. This is because the orbicularis oris muscle response is elicited by hyperexcitable motor neurons after motor neuron inhibition. In addition, indirect orbicularis oris muscle responses should be evident as a waveform with variable latency and amplitude, as observed in the case of the F-wave. However, the latencies of the orbicularis oculi muscle and orbicularis oris muscle responses in the lateral spread tests were consistently present both before and after the diazepam injections. These findings suggest that a direct orbicularis oculi muscle response and indirect orbicularis oris muscle response in the lateral spread test were not elicited by the facial motor neuron. Instead, the direct response was elicited by the orthodromic impulse and the indirect responses were elicited by an impulse that was transmitted to neighboring motor axons at the site of demyelination via axon-axon ephapsis.
Some noteworthy limitations should be discussed to interpret and verify the results of the present experiments. First, the number of subjects included in this study was small; therefore, our results need to be replicated before they can be broadly generalized to all patients with HFS. Second, the difference in the latencies of the R1 reflex and early orbicularis oris muscle response before and after diazepam administration were not large compared with that of the R2 reflex and late orbicularis oris muscle response under the same conditions. This may be because of insufficient motor neuron suppression. However, the R2 reflex and late orbicularis oris muscle response to supraorbital stimulation were markedly delayed, whereas the latency of the orbicularis oris muscle response in the lateral spread test was quite consistent. These findings support our conclusions. Last, we did not assess the amplitude of the responses because we assumed that differences in the latency of responses before and after diazepam injection served as an appropriate indicator of motor neuron excitability.
In summary, we suggest that the approach described here can be used to identify the pathophysiology of HFSs, and we believe that this method is also reliable for evaluating the origin of a lateral spread response.
In conclusion, the orbicularis oris muscle response to supraorbital nerve stimulation in patients with HFS may be caused by increased facial motor neuronal hyperexcitability or the ephaptic response. Stimulating the supraorbital nerve with an electrical stimulus activates the supraorbital nerve afferents that reach the facial motor neurons of the orbicularis oculi muscles. In other words, R1 and R2 responses and the early and late orbicularis oris muscle responses to supraorbital nerve stimulation always pass through the trigeminal and facial motor neurons. Therefore, latency delay of the responses may be observed either in facial motor neuronal hyperexcitability or in ephaptic transmission and ectopic excitation after diazepam administration. Similarly, in the present study, the latency of R1, R2, and early and late responses after supraorbital stimulation was further delayed as time after diazepam administration increased. In contrast, the latency of the orbicularis oris response following stimulation of the zygomatic branch of the facial nerve was quite consistent. These findings suggest that the orbicularis oris muscle response to the lateral spread test is caused by cross-transmission of facial nerve fibers at the site of vascular compression, rather than arising from hyperexcitable facial motor neurons.
Although the central theory of facial motor neuronal hyperexcitability as the underlying pathophysiology of HFS cannot be excluded totally by our current findings, the present data support the peripheral theory that ephaptic transmission contributes to the pathophysiologic mechanism of HFS.


 XML Download
XML Download