

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Idiopathic CD4+ T-lymphocytopenia is defined by the following criteria established by the United States Centers for Disease Control and Prevention in 1992 [1]: 1) a CD4+ T cell count below 300 cells/µL or a CD4+ percentage below 20% occurring on at least two separate laboratory studies, and 2) the absence of concomitant human immunodeficiency virus (HIV) infection or other immunodeficiency. To date, the underlying etiology of idiopathic CD4+ T-lymphocytopenia is not yet understood; moreover, the associated clinical presentation can vary from an asymptomatic state to that of life-threatening severe opportunistic infections [2].
Motor axonal neuropathy is characterized clinically by weakness without sensory loss, with electrophysiological studies revealing decreases in the compound muscle action potential amplitude without evidence of demyelination or sensory involvement, and through pathologic findings indicating macrophage infiltration [3,4]. Many conditions have been associated with motor axonal neuropathy, including Charcot-Marie-Tooth neuropathy type 2, porphyrism, acute motor axonal neuropathy (AMAN, a subtype of Guillain-Barre syndrome), and several toxin-induced neuropathies (hexacarbon inhalation, amiodarone, chloroquine) [4].
This case report describes a rare motor axonal neuropathy associated with idiopathic CD4+ T-lymphocytopenia in the absence of any other cause. While demyelinating polyneuropathies associated with serum immunoglobulin A deficiency [5] and HIV infection [6,7] have been described in the literature, no isolated cases of motor axonal neuropathy have been reported. We identify this atypical case of motor axonal neuropathy associated with idiopathic CD4+ T-lymphocytopenia through laboratory testing as well as neurologic and electrodiagnostic exams; the results are reported here with a literature review.
Go to : 
CASE REPORT
A 10-year-old boy with no significant birth or family history was hospitalized in the pediatric ward of our hospital for recurrent respiratory infections. Per the patient's medical record, he exhibited normal development, though he was hospitalized at 16 months of age for pneumonia. Since that time, the patient was repetitively hospitalized for various infections, including pneumonia, otitis media, and paranasal sinusitis. Given these recurrent infections, an underlying immunodeficiency was suspected. A subsequent laboratory workup was performed, revealing normal serum quantitative immunoglobulin and complement levels as well as a marked decrease in CD4+ T cells. The patient was then diagnosed with idiopathic CD4+ T-lymphocytopenia at the age of 5, given his history of recurrent infections in the setting of a low CD4+ T cell count. All secondary infections were then conservatively treated with antibiotics, and the patient was closely followed as an outpatient, though immunotherapy was not performed.
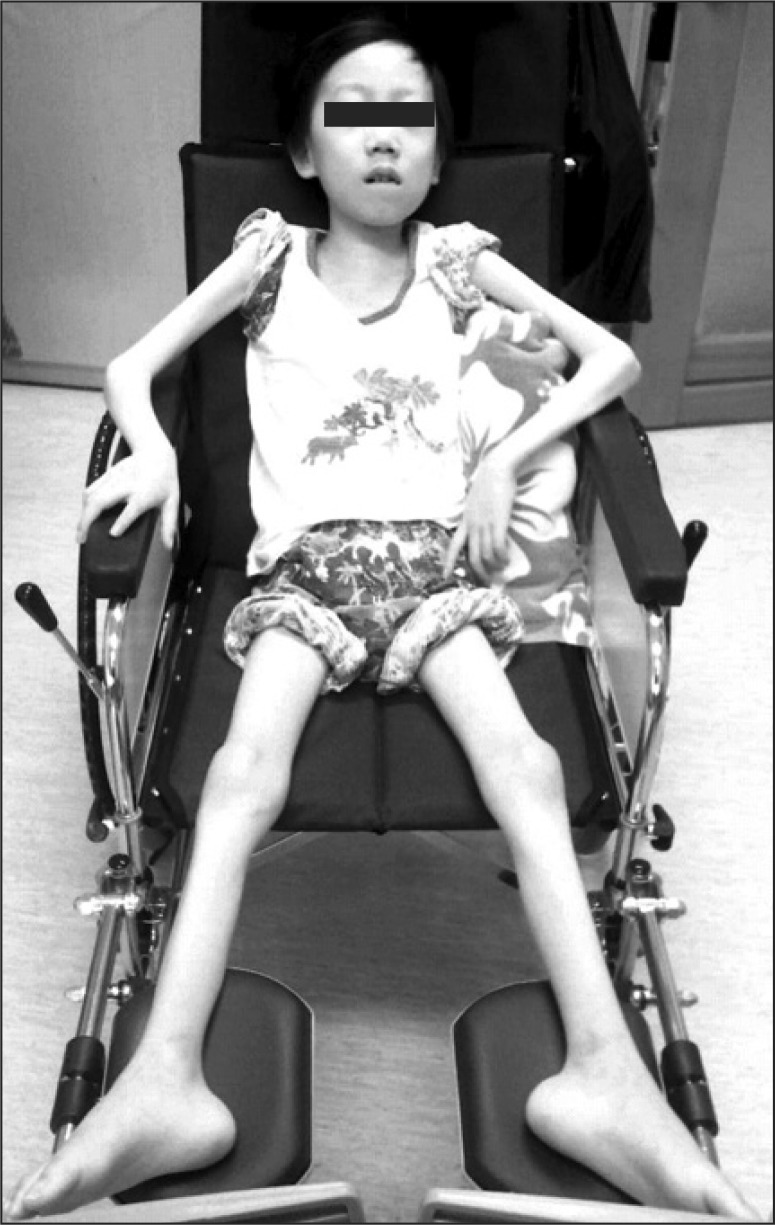
Several years later, in 2010, the patient complained of generalized weakness of his four extremities, although he reported that these symptoms did not significantly affect his daily life. However, as this limb weakness continued to worsen, the patient was then referred to our clinic in the Department of Rehabilitation Medicine in January 2011 for further evaluation and treatment. At the time of his first examination, the patient was 124 cm tall, weighed 19.4 kg, and had a body mass index of 12.36, which is defined as being underweight. Prominent muscular atrophy of the limbs was noted on the exam (Fig. 1). Using the Medical Research Council (MRC) scale, manual muscle testing was then performed revealing the following: upper extremity shoulder flexion 2/5, abduction 2/5, elbow flexion 3/5, extension 2/5, wrist flexion 3/5, and extension 2/5; lower extremity hip flexion 2/5, extension 1/5, knee flexion 1/5, extension 2/5, ankle dorsiflexion 1/5, great toe extension 3/5, and plantarflexion 2/5. All results were symmetric for the contralateral upper and lower extremities. Additionally, no sensory abnormalities were detected in temperature, tactile, position, or vibration sensation. The patient's deep tendon reflexes for the bilateral biceps and knees were also normal. During the entire exam, the patient was conscious and alert, and the cranial nerve examination was without abnormal findings. The patient was able to turn over in bed, but was unable to sit up from a supine position without assistance. In terms of the degree of independence in daily living, the patient had a Modified Barthel Index (MBI) of 28, indicating the need for maximal assistance from other people.
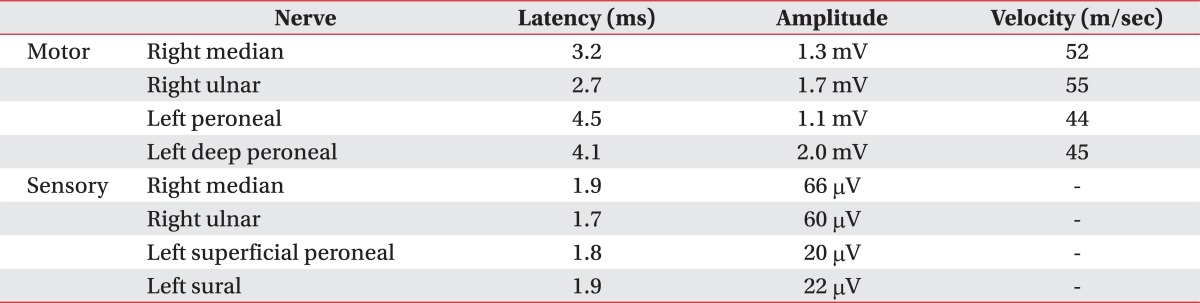
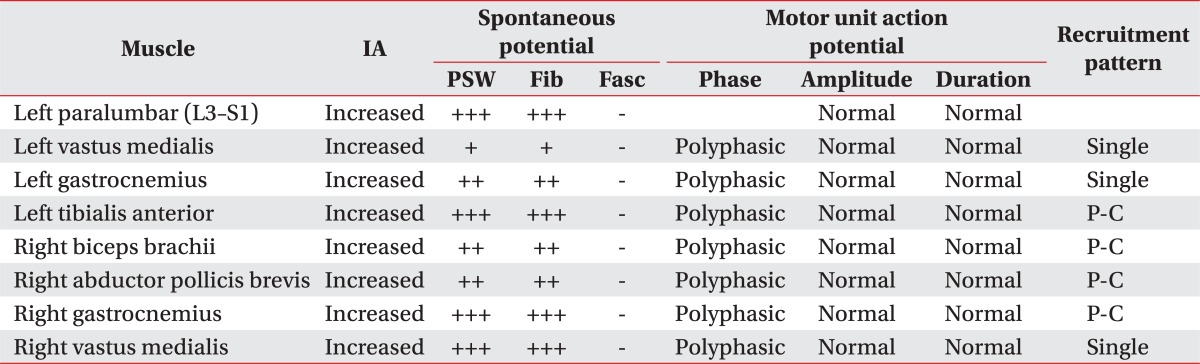
A subsequent motor nerve conduction study showed decreased compound muscle action potential amplitude for the left median, ulnar, right peroneal, and tibial nerves, although the latency and nerve conduction velocity were within normal limits. Additional sensory nerve conduction studies were also within normal limits (Table 1). Needle electromyography further revealed increased insertional activity for all examined muscles, with a large amount of abnormal spontaneous activity noted in the paralumbar muscles, left vastus medialis, tibialis anterior, gastrocnemius, right biceps brachii, and abductor pollicis brevis. Specifically, polyphasic motor unit action potentials and reduced motor unit recruitment were observed during muscle contraction (Table 2). A somatosensory evoked potential study of the tibial nerve; however, it did not reveal any specific findings.
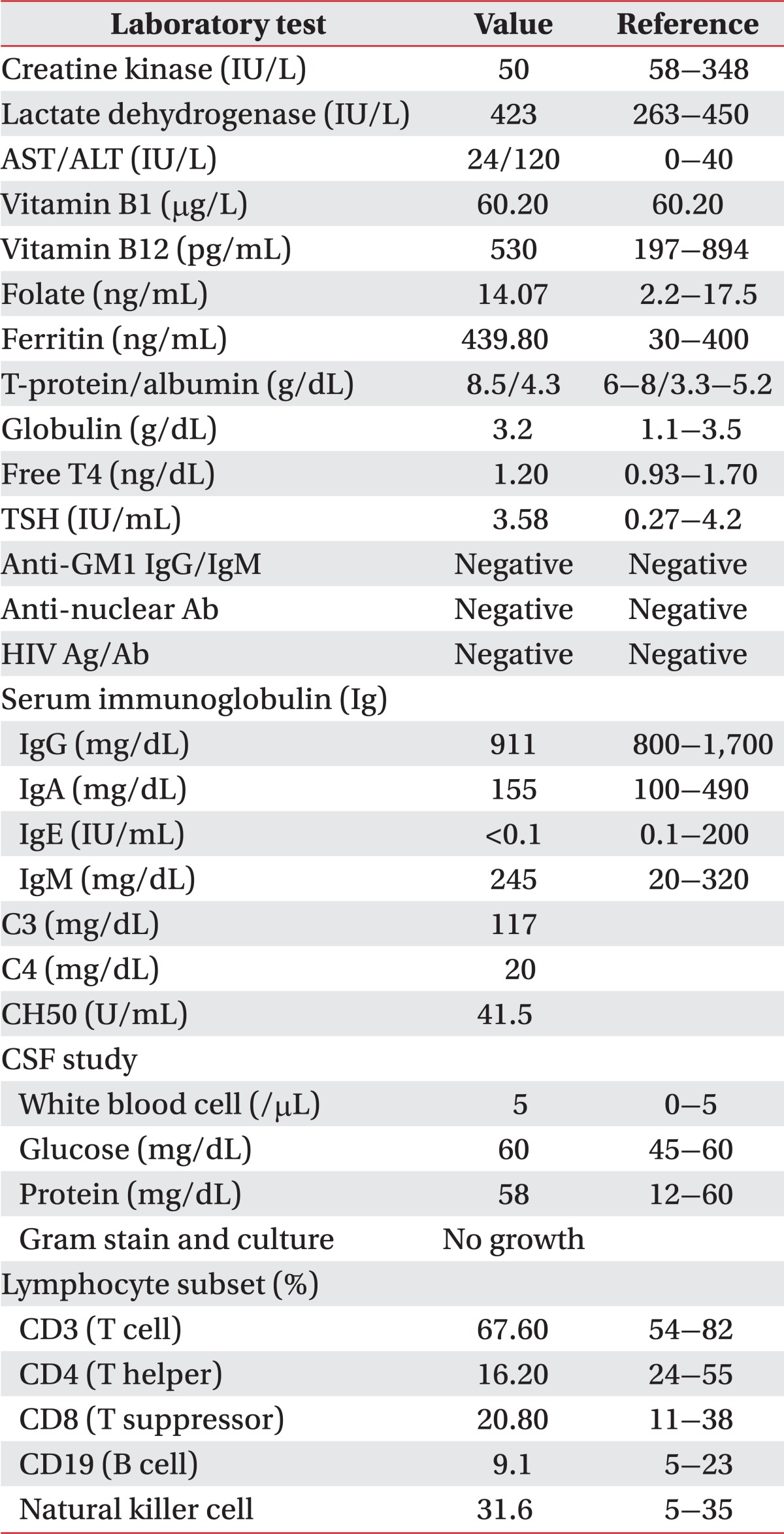
Results from an additional electrodiagnostic study indicated that the motor nerves of the upper and lower extremities were primarily affected, with a marked reduction in compound muscle action potential amplitude in the setting of a normal conduction velocity pattern. The results suggested a major axonal involvement rather than a demyelinating process. A diagnosis of the motor axonal type was then reached, given the results from the sensory nerve studies and lack of associated clinical symptoms. A number of additional diagnostic studies were then performed in order to identify the underlying etiology of the process, with no significant finding noted on the magnetic resonance imaging of the brain, vertebrae, and cerebrospinal fluid (CSF). Serum muscle enzymes, electrolytes, thyroid function tests, and liver function tests were also within the normal range. Additionally, serum immunoglobulin GM1 and GD1 were both negative as was the antibody testing for all associated infectious diseases (e.g., HIV) and abdominal ultrasonography (Table 3). The patient was found to have a decreased number of CD4+ T cells on lymphocyte subtyping (Table 3). A sural nerve biopsy was not performed as the sensory nerve function was found to be normal in the electrodiagnostic study.
Clinically, the patient had significant difficulty in carrying out daily living activities, including walking, due to marked muscle weakness. Rehabilitation treatment focused primarily on wheelchair transfer training, joint contracture prevention, and posture maintenance, since active rehabilitation was not possible due to the patient's recurrent infections. As the pulmonary function testing showed decreased respiratory function, pulmonary rehabilitation was also initiated.
Go to :
DISCUSSION
Decreases in CD4+ T-lymphocytes can occur in a number of settings-including viral and mycobacterial infections, Brucellosis, malnutrition, autoimmune diseases, and acquired immune deficiency syndrome (AIDS)-despite the fact that the associated symptoms are usually temporary and not severe [8]. Conversely, while the exact prevalence of idiopathic CD4+ T-lymphocytopenia is not known, it is estimated to be extremely low, as no cases were identified in a screening study of 2,028 blood donors from 1994 [2]. The clinical manifestations of idiopathic CD4+ T-lymphocytopenia vary from an asymptomatic state to the recurrent severe opportunistic infections similar to those that occur in the setting of AIDS. Although rare cases associated with Sjogren's syndrome, pulmonary sarcoidosis, Down syndrome, and non-Hodgkin's lymphoma exist in the literature, no such reports exist regarding motor axonal neuropathy verified by electrodiagnostic assay.
Idiopathic CD4+ T-lymphocytopenia can likely be induced by a variety of underlying mechanisms, with proposed hypotheses including decreases in T cell progenitors, increases in T cell apoptosis, and antibody production against CD4+ T cells. Neuropathy may also result from similar immunological mechanisms [9]. As noted in idiopathic CD4+ T-lymphocytopenia, decreases in CD4+ T-lymphocytes are also seen in the setting of HIV infection. HIV-associated neuropathy is now believed to be immune-mediated, with cytokine release from macrophages acting as the primary neuronal insult. Other nonimmunological mechanisms include vitamin B12 deficiency and an idiopathic neurotoxic effect from a number of HIV medications [6,7]. Accordingly, the patient described here could have also developed a neuropathy induced by similar immunological mechanisms.
To rule out nonimmunological causes-such as nutritional deficiencies, infectious etiologies, autoimmune disease, and medications-the patient's nutritional status, HIV status, autoimmune antibody status, and history of medication use were thoroughly evaluated. Further, although the patient was underweight (body mass index=12.36), serum protein, albumin, vitamin B1, B12, folic acid, and ferritin levels were all normal, thus, malnutrition-induced neuropathy could be ruled out. When diagnosing idiopathic CD4+ T-lymphocytopenia, HIV infection must also be ruled out; hence, HIV antigen antibody enzyme-linked immunosorbent assay (ELISA) testing was performed four separate times. As all were negative, HIV infection and associated neuropathy were excluded. Moreover, the patient also received multidrug therapy, including many various antibiotics, for recurrent respiratory infections. However, since he had only been treated in the pediatric ward of our hospital, all of the medications administered since birth were documented in his medical record. Specifically, in the past, the patient had been treated with penicillin-class antibiotics, cephalosporins (including first, second, and third generation agents), and carbapenem. At no time were aminoglycosides or sulphonamides, both well-known causes of neuropathy, ever used. Furthermore, as the patient had been intermittently treated with prednisolone, the possibility of an underlying myopathy was also considered. However, as all serum muscle enzyme levels were within normal limits, the motor unit action potential amplitude was within the normal range on needle electromyography, and an early motor unit recruitment pattern was not observed; myopathy was effectively ruled out. Additionally, since the patient did not have a history of intensive care treatment, mechanical ventilation, sepsis, septic shock, or multiple organ dysfunction, critical illness neuropathy-a condition mostly involving both motor and sensory nerves-was also excluded. Other motor neuron diseases (e.g., spinal muscular atrophy) were also excluded for the following reasons: a fasciculation potential was not observed on either of the two performed electromyography, the motor unit action potential duration and amplitude were both within normal limits, the patient's clinical symptoms developed at age 10, bulbar palsy was absent, and the patient's family history was found on the genetic testing.
AMAN, a subtype of Guillain-Barre syndrome, might present as motor axonal neuropathy on the electrodiagnostic study. However, our patient's clinical symptoms were significantly different from AMAN, whereby progress is dramatic and recovery of the muscular strength is rapid. Exclusion of AMAN was then confirmed given the normal CSF testing, negative serum anti-GM1 antibody assay, and lack of therapeutic response to glucocorticoids. Charcot-Marie-Tooth disease type 2 can also manifest as motor axonal neuropathy in a similar fashion to AMAN on the electrodiagnostic study. However, Charcot-Marie-Tooth disease was ruled out by the physical exam (specifically the presence of cavus foot and weakness of the lower limbs) and family history. As described above, a variety of tests were performed in order to identify the underlying cause of the patient's motor axonal neuropathy, all of which were within normal limits, with the exception of the findings specific for idiopathic CD4+ T-lymphocytopenia.
Currently, the therapy for idiopathic CD4+ T-lymphocytopenia focuses on the treatment and prevention of opportunistic infections, although interleukin-2 has been reported as being effective in increasing the CD4+ T-lymphocyte count [9]. Biopsy was not performed in this case, as the patient's parents were unwilling to give consent, which may represent one limitation of this case report. However, a diagnosis of motor axonal neuropathy was nonetheless reached by clinical, physical, and electrodiagnostic examinations.
In conclusion, motor axonal neuropathy secondary to idiopathic CD4+ T-lymphocytopenia is very rare, particularly in a pediatric population. Currently, the exact etiologic mechanisms for these processes are not known, and as such, no causality can be determined. Further studies are necessary to better describe the etiology and immunological characteristics of idiopathic CD4+ T-lymphocytopenia, as well as to establish the diagnostic criteria, the treatment protocol, and further assess the overall prognosis.
Go to :
 XML Download
XML Download