

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The incidence of colorectal cancer (CRC), which is one of the most common cancers and the fourth leading cause of cancer-related mortality in the world, is growing gradually [1]. However, according to previous studies, the pathogenesis of CRC is extremely complex and various factors are known to play a role in its progression from normal mucosa to polyp stage and cancer. In addition to genetic factors, environmental factors such as obesity, lifestyle, chronic diseases of the colon, drugs, and smoking are associated with the development of CRC [234].
Gut microbiota inhabiting the gut microenvironment has been recognized as an important factor in colon cancer development [5678]. In general, 1013 to 1014 gut microbiota exists in the human colon. These balance the immune system and enhance resistance to pathogens by supporting normal physiological function [910]. However, when the normal form of gut microbiota is disrupted due to aging, obesity, diet, antibiotic usage, inflammatory bowel disease, diabetes, or chronic kidney disease, microbial homeostasis is threatened and CRC may develop.
Several previous studies have indicated a link between CRC and the dysbiosis of gut microbiota [111213]. Dysbiosis may cause an increase in the number of harmful microbe populations. These not only produce carcinogens but also cause inflammation by affecting the immune function of intestinal mucosa, in addition to disrupting division of mucosal cells, apoptosis, and chromosomal stability [1415]. Nakatsu et al. [16], reported that microbial communities are mutually exclusive during the progression from normal conditions to CRC, and that microbial populations containing fusobacterium are detected prominently in patients with CRC. Feng et al. [17], analyzed 156 stool samples and classified metagenomic linkage groups via metagenomic sequencing, and identified characteristic microbiome compositions in the stool corresponding to carcinogenesis sequences.
To date, studies have focused on identifying pathogenic microbiota associated with CRC development and clarifying mechanisms underlying tumorigenesis. At present, surgery remains the most effective method available for treating CRC. However, it is unclear whether the microbial community would be altered following curative tumor resection. It was hypothesized that curative surgery would remove the tumor together with the gut microbiota that affected tumorigenesis. To test this hypothesis, we compared the gut microbiota before and after curative surgery and analyzed changes in the intestinal microbial community, postsurgery.
METHODS
Study participants
Clinical stage III CRC patients who attended the National Cancer Center, Korea, between May 2017 and June 2017, were recruited for a prospective pilot study. We excluded the patients with (1) distant metastasis, (2) other bowel disorders such as inflammatory bowel disease, ischemic colitis, and tuberculous colitis, (3) a previous history of bowel resection, and (4) patients on medications that affect gut microbiology. Initially, 12 patients with clinical stage III CRC were recruited. Among these 12, one withdrew his research consent and was excluded. Thus, the remaining 11 patients were enrolled. We collected stool samples from these 11 patients before curative surgery and 6 months after, and analyzed changes in the gut microbial community of each patient. All study procedures were performed in accordance with the Declaration of Helsinki. This study was approved by the Institutional Review Board of the National Cancer Center (NCC2017-0126). Written informed consent was obtained from all patients prior to enrolment.
Stool sampling and DNA extraction
The first set of stool samples was collected prior to the start of bowel preparation for surgery and the second set was collected from the outpatient department 6 months postoperatively. In case the patient had to undergo adjuvant chemotherapy following surgery, stool samples were collected 1 month after chemotherapy.
Fresh stool samples (~30 mL) were collected in OMNIGene GUT tube (DNA Genotek, Ontario, Canada) and brought to the research center within 24 hours. Samples were stored at −70℃ in the laboratory prior to DNA extraction. DNA was extracted using a QIAamp DNA Stool Kit according to the manufacturer's protocol (Qiagen, Valencia, CA, USA). A nanodrop 2000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) was used to check the quality and concentration of the DNA.
Sequencing of 16S rRNA
A 16S rRNA sequencing library was constructed according to the 16S metagenomics sequencing library preparation protocol (Illumina, San Diego, CA, USA) targeting the V3 and V4 hypervariable regions of the 16S rRNA gene. KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, USA) and Agencourt AMPure XP system (Beckman Coulter Genomics, Brea, CA, USA) were used for PCR and purification of PCR products, respectively. The initial PCR was performed with 20 ng template DNA using region-specific primers shown to be compatible with Illumina index and sequencing adapters (forward primer: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′; reverse primer:5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGT ATCTAATCC-3′). Following magnetic bead-based purification of polymerase chain reaction (PCR) products, the second PCR was performed using primers from a Nextera XT Index Kit (Illumina Inc., San Diego, CA, USA) with a limited cycle. Subsequently, purified PCR products were visualized using gel electrophoresis and quantified with a Qubit dsDNA HS Assay Kit Invitrogen (Carlsbad, CA, USA) on a Qubit 3.0 fluorometer. Pooled samples were run on an Agilent 2100 bioanalyzer Agilent Technologies (Santa Clara, CA, USA) for quality analysis prior to sequencing. Libraries were quantified via qPCR using CFX96 Real Time System Bio-Rad Laboratories (Hercules, CA, USA). After normalization, sequencing of the prepared library was conducted on the Miseq system (Illumina) with 300-bp pairedend reads.
Analysis of sequencing data
The reads were sorted using unique barcodes for each PCR product. The barcode, linker, and primer sequences were then removed from the original sequencing reads To merge pairedend reads that were processed in a previous step, FLASH v 1.2.11 software was applied [18]. Merged reads that contained 2 or more ambiguous nucleotides, exhibited a low-quality score (average score < 20), or were shorter than 300 bp, were filtered out. Potential chimeric sequences were detected using the Bellerophon method.
Determination of operational taxonomic units and taxonomic classification
Preprocessed reads from each sample were used to calculate the number of operational taxonomic units (OTUs). The number of OTUs was determined by clustering sequences from each sample via a 97% sequence identity cutoff using QIIME software v1.8.0 (University of Colorado, Boulder, CO, USA) [19]. Taxonomic abundance was evaluated with a RDP Classifier v1.1 (Ribosomal Database Project, Michigan State University Board of Trustees, East Lansing, MI, USA) using a confidence threshold of 0.8 derived from preprocessed reads for each sample. Microbial composition was normalized using the value calculated from the taxonomy abundance count divided by the number of preprocessed reads for each sample.
Statistical analyses
To measure the alpha diversity of each sample, the OTUs were analyzed using the Shannon index as follows: 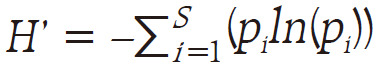 . In order to measure beta diversity, the difference in organism composition was measured according to Bray-Curtis distance as follows:
. In order to measure beta diversity, the difference in organism composition was measured according to Bray-Curtis distance as follows: 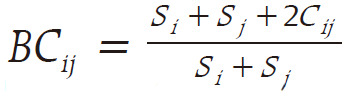 . Principal component analysis (PCA) was then performed using the measured beta diversities. Continuous variables (OTUs and alpha diversity) were analyzed using the Student t-test or Wilcoxon rank-sum test following normality tests. The edge R software version 3.6.8 (Bioconductor, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia) was used to compare the microbial composition between preoperative and postoperative stool samples. An adjusted P-value < 0.05 was considered statistically significant and all statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).
. Principal component analysis (PCA) was then performed using the measured beta diversities. Continuous variables (OTUs and alpha diversity) were analyzed using the Student t-test or Wilcoxon rank-sum test following normality tests. The edge R software version 3.6.8 (Bioconductor, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia) was used to compare the microbial composition between preoperative and postoperative stool samples. An adjusted P-value < 0.05 was considered statistically significant and all statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).
. In order to measure beta diversity, the difference in organism composition was measured according to Bray-Curtis distance as follows: . Principal component analysis (PCA) was then performed using the measured beta diversities. Continuous variables (OTUs and alpha diversity) were analyzed using the Student t-test or Wilcoxon rank-sum test following normality tests. The edge R software version 3.6.8 (Bioconductor, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia) was used to compare the microbial composition between preoperative and postoperative stool samples. An adjusted P-value < 0.05 was considered statistically significant and all statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).RESULTS
Subject characteristics
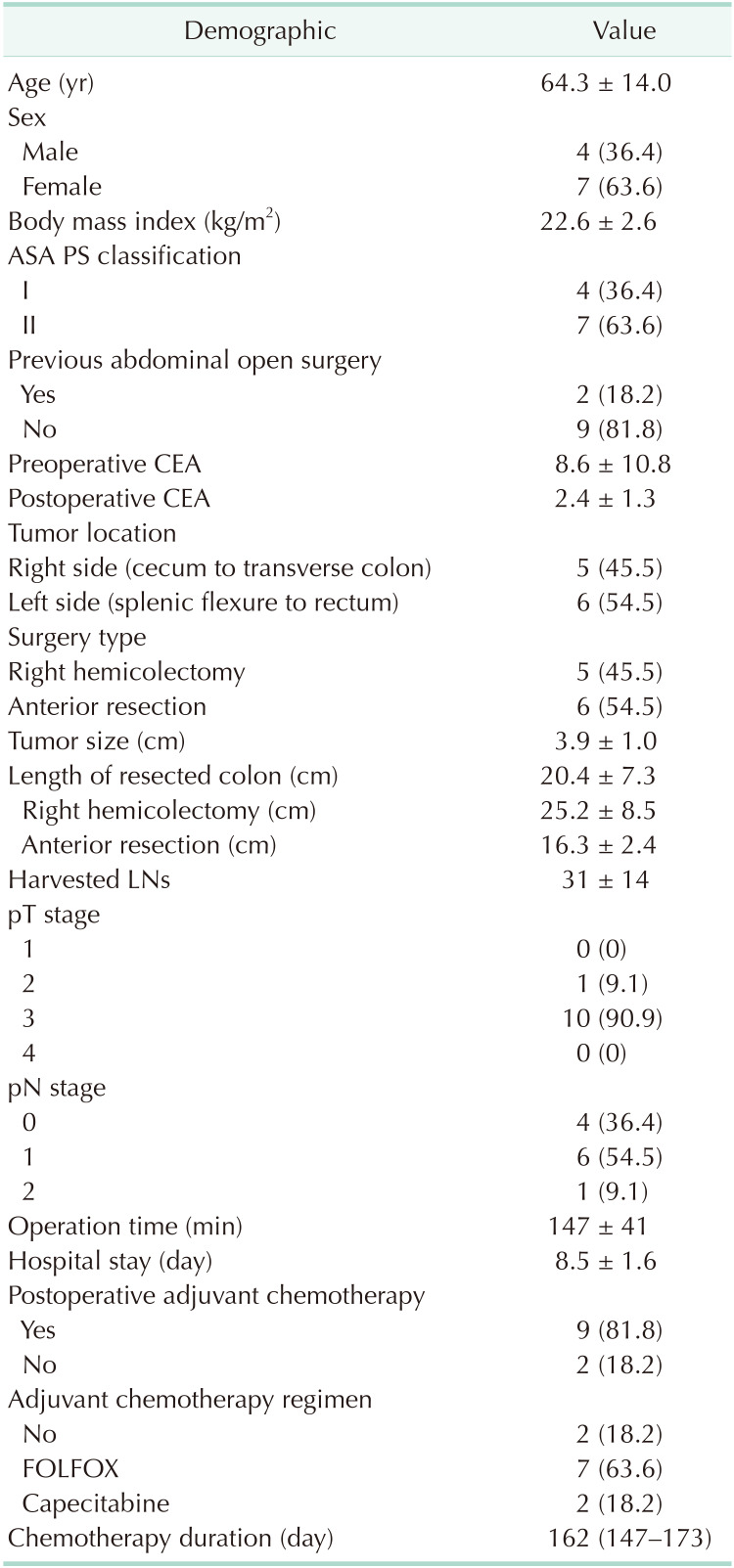
The demographic and clinical characteristics of the participants are shown (Table 1). Mean age was 64.3 ± 14.0 years and 7 patients (63.6%) were female. Tumors were evenly located; as 5 patients exhibited right side (cecum to transverse colon) tumors and 6 patients exhibited left side (splenic flexure to rectum) tumors. All surgeries were performed via a laparoscopic procedure. Right hemicolectomy and anterior resection were performed according to the location of the lesion. Nine patients underwent postoperative chemotherapy. Among these, 7 received FOLFOX and 2 received Capecitabine. FOLFOX (1 cycle; oxaliplatin, 85 mg/m2, LV 200 mg/m2 bolus 5-FU 400 mg/m2 on day 1, infusional 5-FU 1200 mg/m2 on day 1 and day 2) and Capecitabine (1 cycle; 1,000 mg/m2, oral, twice daily, from day 1 to day 14) treatments were followed by the standardized regimen. Twelve cycles of FOLFOX with 2-week intervals in between each cycle and 8 cycles of Capecitabine with 3-week intervals in between, were conducted. Median duration of chemotherapy was 162 days (147–173 days). All patients completed chemotherapy until the last cycle, but 3 received reduced FOLFOX chemotherapy due to chemotherapy toxicity.
Microbial diversity across all samples
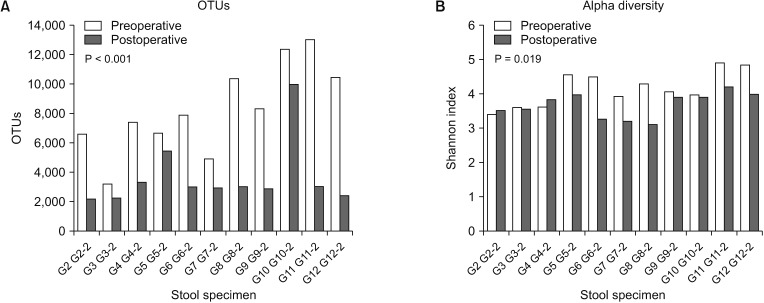
In all patients, the number of OTUs decreased following surgery. The median number of OTUs in postoperative stool samples was significantly smaller (2,981; range, 2,195–10,002) than that (7,920; range, 3,201–13,025) in the preoperative stool samples (P < 0.001) (Fig. 1A). In most patients (9 of 11), alpha diversity (Shannon Index) decreased after surgery. We found that mean alpha diversity of postoperative stool samples was also significantly lower (3.68 ± 0.37) than that (4.16 ± 0.51) in preoperative stool samples (P = 0.019) (Fig. 1B). However, no differences in postoperative alpha diversity were found between right hemicolectomy (3.48 ± 0.31) and anterior resection (3.86 ± 0.34) (P = 0.085) (Supplementary Fig. 1).
The PCA plot for beta diversity showed a differential pattern between preoperative and postoperative stool samples (Fig. 2).
Comparison of fecal microbial composition
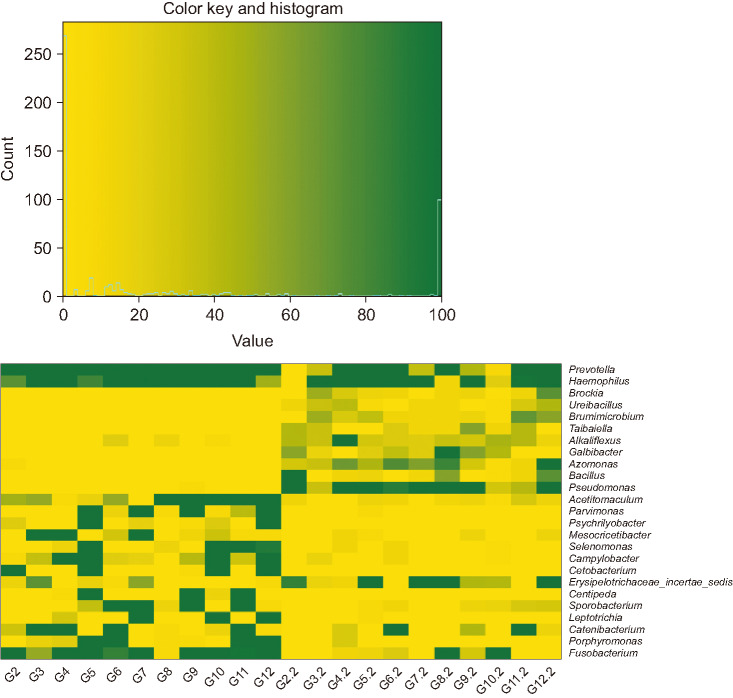
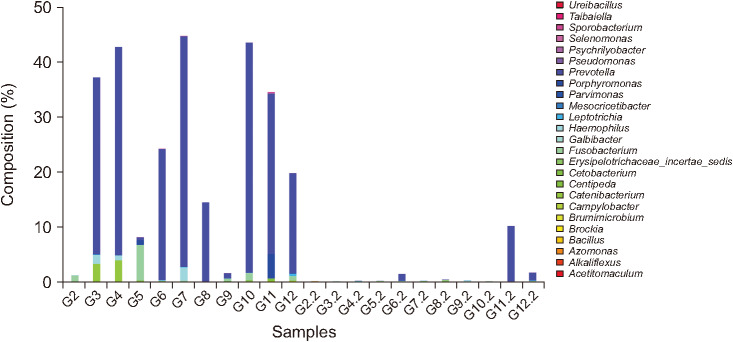
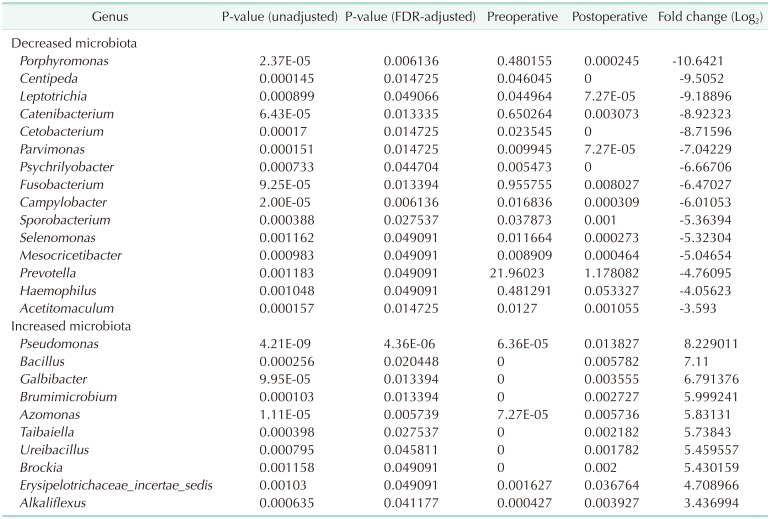
The list of taxa showing significant differences in abundance between preoperative and postoperative gut microbiota at the genus and phylum level, respectively are shown (Tables 2, 3). At the genus level, the abundances of 15 taxa were decreased while those of 10 taxa were increased. The proportions of previously known pathogens, such as Campylobacter (preoperative vs. postoperative 0.0168% vs. 0.0003%; adjusted P = 0.006), Fusobacterium (0.9557% vs. 0.0080%; adjusted P = 0.013), Haemophilus (0.4813% vs. 0.0533%; adjusted P = 0.049), Porphyromonas (0.4802% vs. 0.0002%; adjusted P = 0.006), and Prevotella (21.9602% vs. 1.1781%; adjusted P = 0.049) were significantly decreased following surgery. At the phylum level, the proportion of Fusobacteria (preoperative vs. postoperative 0.0103% vs. <0.0001%; adjusted P < 0.001) was significantly decreased following surgery. The changes in microbial composition of preoperative and postoperative stool samples at the genus level are shown (Figs. 3 and 4). These showed that the pathogens including Prevotella and Fusobacterium were significantly reduced after surgery (Fig. 4).
DISCUSSION
In this study, we compared the gut microbiota in CRC patients before and after curative surgery. OTUs and alpha diversity were significantly decreased following curative surgery. The composition of several bacterial taxa was changed after surgery. The proportions of known pathogens including Campylobacter, Fusobacterium, Haemophilus, Porphyromonas, and Prevotella were decreased at the genus level, while the proportion of Fusobacteria was decreased at the phylum level. Thus, our results proved the hypothesis that pathogens causing carcinogenesis would be decreased following tumor removal via surgery.
Several studies have reported that gut microbiota predominate in the fecal samples of CRC patients [202122]. Fusobacterium, in particular, is one of the most important pathogens which are reported to be closely associated with CRC prognoses [23]. Our study indicated that, following curative resection, Fusobacterium was significantly decreased at both genus and phylum levels. Recently, one pilot study reported changes in gut microbiota 1 month after palliative surgery or radical surgery in patients with CRC [24] where, consistent with our results, alpha diversity decreased after surgery. In that study, pathogens such as Fusobacterium and Parvimonas decreased at the genus level following surgery, but Klebsiella, which often predicts gut flora dysbiosis, increased. They explained that surgery itself may weaken the community's ability to resist pathogens. However, our study did not show an increase in opportunistic pathogens, including Klebsiella. This may be because our study analyzed stool samples 6 month after surgery, by which time the patient would have fully recovered from a stressful surgical condition.
Similarly, Jin et al. [25], reported that postoperative CRC patients showed different microbiota at the genus level than colorectal patients who had not undergone surgery. However, their study was a cross-sectional study, wherein preoperative and postoperative stool samples were not collected from the same CRC patients. This raised the possibility of microbiota changes being caused by clinical variables other than surgery. Our study, on the contrary, was able to reduce confounding factors by performing pairwise comparisons in the same group. Kong et al. [26], reported that, although tumor-related bacteria in patients who received adjuvant capeOx therapy decreased after radical CRC surgery, some conditional pathogens increased. These results may be due to the chemo-agent having a greater effect on the microenvironment than the surgical effect, because of analyzed stool samples being collected between chemotherapy cycles. In our study, 9 out of 11 patients received postoperative chemotherapy and 7 patients received FOLFOX. Stool samples were collected one month following chemotherapy from patients who received postoperative chemotherapy. Previous studies have reported that the microbial environment could recover approximately 1 week following the administering of agents such as antibiotics or chemotherapy [2728]. Therefore, changes in gut microbiota due to chemotherapy are expected to be minimal and no conditional pathogens reported in previous studies were found in our study.
Studies have reported that CRC associated dysbiosis did not induce significant changes in the diversity or abundance of microbial communities [1729]. However, several studies have reported reduced diversity in postoperative patients [242530]. It is known that the use of antibiotics dramatically reduces the diversity of gut microbiota [27]. Also, chemo-agents which induce mucositis in CRC chemotherapy patients may significantly change the diversity and abundance of gut microbiota. In our study, stools were sampled 6 months following surgery, and therefore, decreased diversity could not be attributed to antibiotics that were used during surgery. At our institute, postoperative chemotherapy is performed approximately 1 month after surgery for CRC, and stool samples were collected one month after chemotherapy from patients who received postoperative chemotherapy. Therefore, in patients (9 out of 11) who received postoperative chemotherapy, a second sample was collected approximately 8 months after surgery. Whether the chemotherapy and the different times of second stool collection affect the microbiota is unclear because of the small number of cases. Large-scale prospective studies are needed to further assess this issue.
The study had some inherent limitations. Firstly, the small sample size may not be sufficient to clarify and validate the effects of curative surgery on gut microbiota. The same reason made it difficult to analyze the relationship between gut microbiota and clinical variables. Although age, sex, body mass index, and location of tumor may all have an effect on microbial environment, we were unable to analyze the effect of those factors due to small sample size. Secondly, the association between changes in gut microbiota and prognosis was not analyzed. The current study did not include long-term patient follow-up, and therefore could not clarify whether postoperative microbiota changes were correlated with tumor recurrence.
However, this pilot study provides some information regarding changes in gut microbiota following curative resection of lesions; as such, changes in gut microbiota before and after surgery may be expected to play a role as predictors of treatment efficacy and cancer prognosis.
In conclusion, significant changes in intestinal microbial communities were noted after curative resection of CRC. Especially, pathogenic bacterial populations such as Fusobacterium and Prevotella, which are known to be associated with CRC development, were found to be decreased even though OTUs and alpha diversity were also decreased following curative resection. To determine and validate the clinical significance of these findings, large scale, prospective studies that include cancer prognoses are felt to be mandatory.


 XML Download
XML Download